 |
| Temas de FC |
E. Frauca Remacha![]() , M. García Vega
, M. García Vega
Servicio de Hepatología y Trasplante Hepático Infantil. Hospital Universitario La Paz. Madrid
| Resumen
La incidencia de los tumores hepáticos durante la infancia es baja. De entre los tumores benignos, el más frecuente es el hemangioma, mientras que el hepatoblastoma y el hepatocarcinoma son los tumores malignos más frecuentes. En la mayoría de los casos, se va a detectar el tumor tras una ecografía, bien tras palpación de una hepatomegalia o bien en el contexto de un examen rutinario. El hemangioma presenta un ciclo natural de crecimiento y posterior involución, por lo que el tratamiento solo está indicado en aquellos casos sintomáticos. En los casos de un hepatoblastoma o hepatocarcinoma, la curación de la enfermedad pasa por la resección completa del tumor y tratamiento con quimioterapia adyuvante. La supervivencia de los niños con tumores hepáticos, especialmente en el caso del hepatoblastoma, ha mejorado significativamente tras la introducción de pautas de quimioterapia efectivas y definición de estrategias quirúrgicas, incluyendo el trasplante hepático, apropiadas en cada caso. |
| Abstract
The incidence of liver tumors during childhood is low. Among benign tumors, the most common is hemangioma, while hepatoblastoma (HB) and hepatocarcinoma (HCC) are the most common malignant tumors. In most cases, the tumor will be detected after an ultrasound, either after palpation of hepatomegaly or in the context of a routine examination. Hemangioma presents a natural cycle of growth and subsequent involution, so treatment is only indicated in symptomatic cases. In cases of hepatoblastoma or hepatocarcinoma, the cure of the disease involves complete resection of the tumor and treatment with adjuvant chemotherapy. The survival of children with liver tumors, especially HB, has improved significantly after the introduction of effective chemotherapeutic regimens and appropriate surgical approaches, including liver transplantation, resulting in an increase in the number of patients undergoing definitive tumor resection and a decrease in the incidence of postsurgical recurrences. |
Palabras clave: Tumores hepáticos; Diagnóstico; Opciones terapéuticas; Supervivencia; Recurrencia.
Key words: Liver tumors; Diagnosis; Therapeutic options; Survival; Recurrence.
Pediatr Integral 2025; XXIX (1): 49 – 57
OBJETIVOS
• Conocer los distintos tipos de tumores hepáticos en niños.
• Conocer las variantes clínico-patológicas de cada tipo tumoral.
• Exponer los modos de presentación clínica y métodos diagnósticos.
• Presentar las diferentes posibilidades terapéuticas y el pronóstico de los tumores más frecuentes.
Tumores hepáticos
https://doi.org/10.63149/j.pedint.7
Introducción
Los tumores hepáticos infantiles son tumores relativamente infrecuentes, con una incidencia estimada de entre 0,5-2,5 casos por millón de habitantes. Suponen, por otro lado, del 0,5 al 2 % de todas las neoplasias infantiles y el 4 % de los tumores sólidos en niños. De entre los diferentes tipos de tumores hepáticos que se diagnostican con cierta frecuencia en pacientes pediátricos (Tabla I), el hepatoblastoma, hepatocarcinoma y el hemangioma hepático infantil suman dos tercios de todos los casos aproximadamente(1,2).

Tumores benignos
Hemangioma hepático infantil
Es el tumor hepático benigno más frecuente y un 90 % de los casos se diagnostican en los primeros meses de vida. Las pruebas de imagen son la base para el diagnóstico y solamente está indicado el tratamiento en aquellos casos sintomáticos.
Representa el tumor hepático más frecuente durante el primer año de vida y, de entre los benignos, el más frecuente en la infancia. Entre un 80 % y 90 % de los casos se manifiestan en los dos primeros meses de vida. Presenta un similar comportamiento biológico al del resto de hemangiomas infantiles de otras localizaciones, como la piel. Es importante su distinción de otras lesiones diferentes, como son el “hemangioma hepático del adulto” (malformación vascular que no involuciona) o del “hemangioendotelioma epitelioide” (tumor maligno con capacidad metastásica y excepcional en la infancia).
Es un tumor vascular benigno compuesto predominantemente por células endoteliales y pericitos, que carece de zonas sólidas sarcomatosas y de la capacidad de invasión vascular (sinusoides, venas hepáticas o portales). Pueden ser lesiones únicas (55 %) o múltiples (45 %), afectando a uno o a los dos lóbulos hepáticos. Comparten con el resto de angiomas infantiles la existencia de tres fases en su ciclo vital: a) fase de alta proliferación y rápido crecimiento; b) fase de involución espontánea, con una duración de entre 5 y 10 años, en la que los fenómenos de apoptosis predominan sobre las mitosis, y se produce un reemplazo progresivo de las células endoteliales y pericitos características de la primera fase por estroma; y c) fase de tumor involucionado de forma definitiva, donde todo el tejido original ha sido sustituido por otro fibroso y graso.
En función del momento temporal de su desarrollo, la ISSVA (Sociedad Internacional para el Estudio de Anomalías Vasculares) define dos subtipos:
1. Hemangioma congénito con inicio de la proliferación intraútero y desarrollo completo en el momento del nacimiento. En su involución se distinguen tres patrones: rápida en <2 años (RICH), parcial (PICH) y no involución (NICH).
2. Hemangioma infantil con inicio de proliferación al nacimiento y hasta los 6-12 meses. Involución progresiva hasta los 3-9 años.
Por otro lado, una segunda clasificación, con base en características de imagen, anatomopatológicas y en su comportamiento, diferencia tres subtipos macroscópicamente diferentes: focal, multifocal y difuso (Fig. 1).
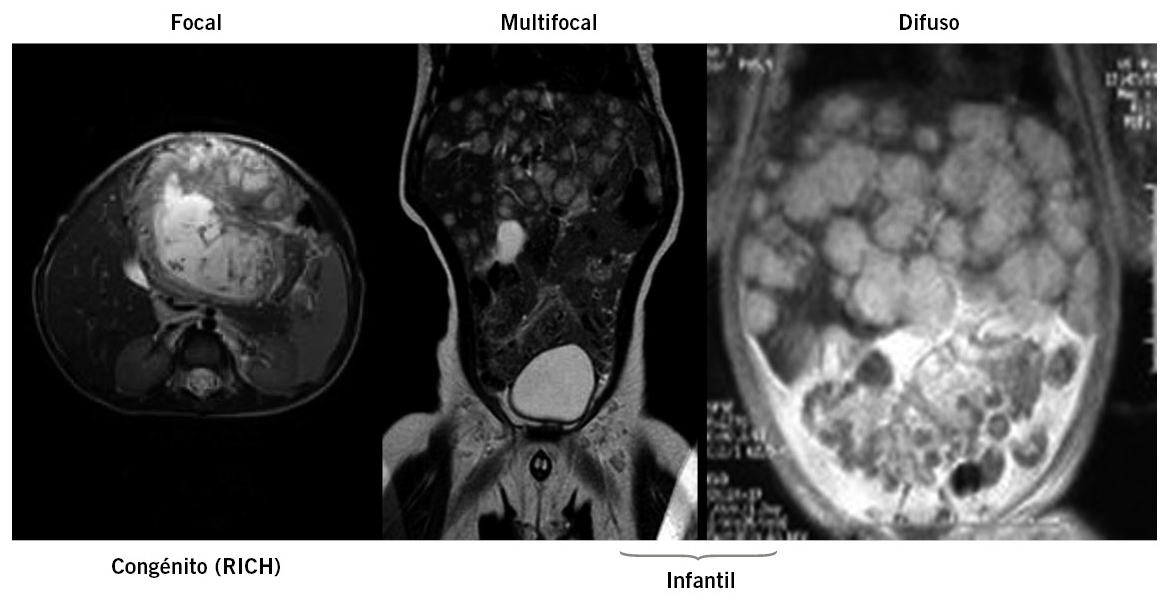
Figura 1. Hemangioma hepático: 3 patrones. Subclasificación 2007 (criterios radiológicos, comportamiento biológico). RICH: patrón de involución rápida en <2 años.
Las lesiones focales son únicas, bien definidas, esféricas, y frecuentemente presentan áreas de necrosis o hemorragia central. En la RMN aparecen como lesiones hipointensas en T1 e hiperintensas en T2 respecto al parénquima hepático circundante. En la mayoría de los casos, se trata de lesiones asintomáticas, muchas de ellas descubiertas accidentalmente en una ecografía prenatal, y que involucionarán rápidamente en la mayoría de los casos (RICH), aunque están descritos casos con insuficiencia cardiaca (ICC) de alto gasto al nacimiento. El subtipo multifocal, cuyas múltiples lesiones esféricas presentan las mismas características radiológicas que las lesiones focales, es también asintomático en la mayoría de los casos, aunque también algunos pacientes pueden presentar una ICC debido a la presencia de shunts arterio o veno-venosos. En la mayoría de ocasiones, asocian hemangiomas cutáneos y, al igual que estos, presentan positividad inmunohistoquímica para el marcador endotelial GLUT-1 y un proceso involutivo similar de las lesiones. Por último, las lesiones difusas (hemangiomatosis hepática difusa) son aquellas que afectan a la práctica totalidad del parénquima hepático con incontables lesiones. Estos casos son, generalmente, clínicamente más severos debido a la gran hepatomegalia que presentan, con compromiso respiratorio por compresión torácica y de la cava inferior e incluso, en algunos casos, con desarrollo de un síndrome compartimental abdominal. Asocian, además, en un porcentaje de casos, un hipotiroidismo severo debido a la producción de iodotironina deiodinasa que inactiva la hormona tiroidea. Por el contrario y a diferencia de los tumores multifocales, y a pesar de su gran “masa tumoral”, es muy infrecuente que asocien una insuficiencia cardiaca de alto gasto(3).
Microscópicamente, se diferencian dos patrones distintos, definidos por Dehner e Ishak: tipo I (más frecuente), con canales vasculares dilatados delimitados por una única capa de células plumosas endoteliales sobre un estroma fibroso; y tipo II (aproximadamente un 20 %), que consiste en canales vasculares con ramificaciones irregulares delimitados por células endoteliales pleomórficas e hipercromáticas, a veces, en acúmulos. En ocasiones, un mismo tumor puede contener ambos patrones histológicos. En un 50 % de los casos, pueden verse grandes canales vasculares que recuerdan al hemangioma cavernoso.
Es fundamental, desde un punto de vista pronóstico y terapéutico, diferenciar este tipo de tumor de un angiosarcoma. No siempre es sencillo e incluso se han descrito casos con rasgos anatomopatológicos de ambos tipos tumorales coexistentes. No existe acuerdo en cuanto a si la presencia de un patrón tipo II representa una forma de angiosarcoma o, más probablemente, un simple cambio degenerativo del tumor. Se ha sugerido, como conveniente, un elevado índice de sospecha de lesiones malignas sarcomatosas en aquellos tumores vasculares de aparición tardía (por encima de los tres años de edad).
El debut clínico se produce habitualmente en los primeros meses de vida (un tercio aproximadamente durante el primer mes) y con discreta preferencia por el sexo femenino. En la mayoría de los casos sintomáticos, son la distensión o la palpación de masa abdominal los signos clínicos iniciales. Según datos del Hepatic Hemangioma Registry, sobre 123 casos de hemangioma multifocal o difuso, un 24 % desarrollaron ICC y un 33 % hipotiroidismo. Otros síntomas posibles son: escasa ganancia ponderoestatural, fiebre y, de forma excepcional, pueden presentar anemia/trombopenia por sangrado intratumoral o intraperitoneal, ictericia o fallo hepático. No es infrecuente la coexistencia de otras localizaciones tumorales, preferentemente en la piel (70 %), pulmonares, encéfalo u otros órganos. Está descrita una tasa de mortalidad en caso de tumores multifocales/difusos entre un 4 % y un 16 %.
No existe un patrón analítico característico, habiéndose señalado la anemia (50 %), hiperbilirrubinemia (20 %) y elevación de transaminasas (32 %) como alteraciones más frecuentes. La cifra de alfa-fetoproteína en suero no está elevada para la edad que presentan la mayoría de estos pacientes, ya que se consideran normales hasta los seis meses de edad cifras posteriormente consideradas como elevadas.
Debido al elevado riesgo de sangrado que comporta la biopsia de estos tumores, el diagnóstico se realiza habitualmente en función de la combinación de rasgos clínicos (masa abdominal en lactante de menos de 3-6 meses, coexistencia de angiomas cutáneos…) y pruebas de imagen (ecografía Doppler, tomografía, angio-resonancia). Solamente debe asegurarse el diagnóstico mediante biopsia en aquellos casos de presentación “atípica”, como tener edad mayor de un año o mostrar signos radiológicos diferentes a los habituales (lesión homogénea sólida con refuerzo centrípeto). En los casos con shunts de alto flujo, es habitual y característica la reducción del calibre de la aorta distal al tumor por el alto grado de “robo” vascular que ocasiona.
No existe actualmente una pauta de tratamiento universalmente aceptada. En general, la decisión de tratamiento y la modalidad de este dependen de la presencia o no de síntomas al diagnóstico y del subtipo de lesión. En el caso de que el tumor sea focal o multifocal y asintomático, es razonable una actitud expectante en espera de la probable regresión tumoral. Por el contrario, en aquellos de estos casos con ICC, se ha demostrado necesario el tratamiento con fármacos con actividad antiangiogénica que acelere la involución tumoral, además de tratamiento de soporte con digoxina y diuréticos. Se añadirá tratamiento con hormona tiroidea en casos de hipotiroidismo(4,5).
Actualmente, y en función de la eficacia en el tratamiento de angiomas cutáneos, se usa el propranolol (2 mg/kg/día oral) con muy buenos resultados (reducción del tamaño del tumor y mejoría/desaparición de los síntomas) y apenas efectos secundarios, por lo que representa la primera opción terapéutica en tumores multifocales y difusos(6,7). Los corticoides (prednisona 2-3 mg/kg/día) representan la alternativa al propranolol en este tipo de tumores y en casos en los que esté contraindicado. Por el contrario, en tumores focales sintomáticos, es el tratamiento de elección.
Alternativamente, se pueden contemplar otras opciones de tratamiento en casos seleccionados, como la resección/hepatectomía de lesión única accesible quirúrgicamente; la embolización de arteria hepática, en casos con un shunt de alto flujo e insuficiencia cardiaca; o incluso, el trasplante hepático en casos de angiomatosis difusa, en los que, a pesar de todas las demás medidas de tratamiento disponibles, el paciente permanece con síntomas y elevado riesgo vital.
Hamartoma mesenquimal
Segundo tumor benigno más frecuente. Diagnóstico habitual en niños menores de 2 años. En algunos casos, el tumor regresa espontáneamente, pero, en la mayoría de los casos, está indicada la resección.
Tras el hemangioma, es el segundo tumor benigno hepático más frecuente y representa entre el 6-8 % de todas las neoplasias hepáticas infantiles. Se presenta de forma típica antes de los 2 años de edad e incluso se han diagnosticado casos intraútero mediante una ecografía fetal rutinaria.
El primer síntoma suele ser la palpación de una masa hepática y, en fases más avanzadas, el crecimiento tumoral, que puede ser rápido, puede provocar compresión de la vena cava inferior, dificultades para la alimentación o incluso compromiso respiratorio. Se trata habitualmente de una neoplasia única y grande, localizada con más frecuencia en el lóbulo hepático derecho. Las pruebas de imagen (ecografía, TC) revelan una masa bien delimitada, poliquística, que contiene quistes de densidad baja, separados por tabiques sólidos y estroma, aunque en ocasiones, cuando dichos quistes son de pequeño tamaño, la apariencia del tumor es sólida y puede sugerir un tumor maligno, por lo que será necesaria la biopsia para el diagnóstico diferencial. Histológicamente, son tumores compuestos de células endoteliales mezcladas con tejido fibroso y mixoide, formando áreas sólidas y quísticas.
La evolución habitual en el tiempo es el crecimiento del tumor durante los primeros meses de vida para posteriormente estabilizarse, continuar creciendo o regresar espontáneamente. Esta última posibilidad sustenta la opinión de no extirpar quirúrgicamente aquellos tumores asintomáticos frente a la más extendida de resección en todos los casos, sintomáticos o no, teniendo en cuenta la posibilidad, aunque infrecuente, de malignización de este tumor hacia un sarcoma embrionario. Se han publicado casos aislados de trasplante hepático en este tipo de tumor, quedando reservado como opción terapéutica para aquellos casos en que la resección quirúrgica no sea posible o fracase.
Otros tumores benignos
• Adenoma hepático. Tumor raro de incidencia mayoritaria en mujeres jóvenes en tratamiento con anticonceptivos. El tratamiento es la resección o la ablación con radiofrecuencia.
• Teratoma hepático. Muy infrecuente. Los escasos casos reportados muestran una incidencia predominante en niños menores de tres años. En estos casos, se describen rasgos de malignidad en casi la mitad de los mismos, por lo que deben ser resecados.
Tumores malignos
Hepatoblastoma
Es el tumor hepático maligno más frecuente en la infancia y el 90 % de los casos se diagnostica en los primeros cinco años de vida. El diagnóstico de sospecha se basa en una prueba de imagen y lo confirma la biopsia del tumor. La curación pasa por la resección mediante hepatectomía o trasplante hepático y quimioterapia coadyuvante.
El hepatoblastoma es el tumor hepático maligno más frecuente en la infancia y supone, aproximadamente, el 1 % de todos los tumores pediátricos y alrededor del 75 % de los cánceres hepáticos en niños. Se le calcula una incidencia anual en los países occidentales de entre 0,5 y 1,5 nuevos casos por millón de niños menores de quince años. Afecta, fundamentalmente, a niños entre los 6 meses y 3 años de edad (90 % se diagnostica en los primeros 5 años de vida), aunque también han sido comunicados casos entre neonatos o adolescentes.
No se ha descrito la predilección racial del tumor y sí un predominio masculino de entre 1,5:1 a 2:1 sobre el sexo femenino. Como factores de riesgo, se han reseñado la prematuridad o el peso bajo al nacimiento (<1.000 g), lo que podría ser la razón del aumento de incidencia del tumor en la última década.
Aunque la gran mayoría son esporádicos, se han comunicado casos familiares de hepatoblastomas asociados a entidades, como la poliposis adenomatosa familiar y el síndrome de Beckwith Wiedemann, lo que sugeriría un cierto papel en la patogenia de estos tumores de los cromosomas 5 y 11, respectivamente. El análisis de las células de este tipo de tumor no ha mostrado un patrón determinado de anomalías citogenéticas. En general, se asume que los casos esporádicos de hepatoblastoma representan un tipo de tumor heterogéneo desde un punto de vista molecular. No se han establecido los mecanismos patogénicos responsables, aunque parece cobrar protagonismo la hipótesis de un incremento en la producción de un factor de crecimiento hepático por parte de fibroblastos y células endoteliales, tras el incremento en la producción de determinadas interleukinas por parte de las células tumorales. Se ha sugerido la posible inactivación de genes supresores tumorales (como el gen APC en cromosoma 5 que regula la vía beta-catenina o el p53), como posible mecanismo implicado en la patogenia tumoral.
Es un tumor embrionario en el sentido de presentar una diferenciación incompleta y derivarse de células stem hepáticas, con la potencialidad de convertirse en hepatocitos o células del epitelio biliar. Existen dos subtipos: a) tipo “epitelial”, con tejido epitelial predominante con células embrionales, fetales o ambas; y b) tipo “mixto”, con tejido mesenquimal (cartílago y osteoide) añadido a los elementos epiteliales. Hoy en día, se reconocen, a su vez, cinco subtipos “epiteliales” en función del grado de diferenciación celular: a) “fetal”, muy diferenciado, recordando sus células a hepatocitos maduros (31 %); b) “embrional”, pobremente diferenciado (19 %); c) “anaplásico” o indiferenciado de células pequeñas (3 %); d) “macrotrabecular”, similar al hepatocarcinoma (3 %); y e) “colangioblástico”. En referencia al pronóstico, parece el tipo “fetal” puro como el más favorable; se asocia la indiferenciación histológica con un pronóstico desfavorable y en el tipo “mixto” una mayor presencia de tejido mesenquimal parece mejorar el pronóstico.
El debut clínico habitual es el descubrimiento de una masa abdominal asintomática en un niño pequeño. En estadios avanzados pueden presentar, además: pérdida de peso, anorexia, vómitos y dolor abdominal. Un 5 % presenta ictericia. En la exploración física se objetiva una masa firme e irregular en cuadrante superior derecho del abdomen y que, en ocasiones, cruza la línea media del mismo. Un 20 % de los pacientes presenta metástasis en el momento del diagnóstico, en la mayoría de los casos de localización pulmonar, aunque también se han descrito en el sistema nervioso central y en otras localizaciones. No son frecuentes las metástasis óseas ni la afectación de la médula ósea.
Entre los hallazgos de laboratorio destacan la anemia (70 %), trombocitosis (50 %) y, por encima de todas ellas, la elevación marcada de las cifras de alfa-fetoproteína (90 % de los pacientes), que la convierten en un marcador muy sensible, aunque inespecífico (puede aumentar en cualquier enfermedad hepática que asocie regeneración del hígado), para el diagnóstico. Asimismo, es muy útil como marcador de la respuesta al tratamiento y de la recurrencia tumoral. Es importante recordar que la alfa-fetoproteína en condiciones normales puede alcanzar niveles de hasta 25.000-50.000 ng/ml al nacimiento y no desciende hasta los niveles “adultos” de menos de 25 ng/ml hasta los 6 meses de vida.
Las técnicas de imagen juegan un papel de suma importancia tanto en el diagnóstico y estadiaje de estos tumores como en la decisión y seguimiento de su tratamiento. La ecografía es con frecuencia el medio diagnóstico inicial con la objetivación de una (o varias) masa bien definida, intrahepática, hiperecoica, sólida, no encapsulada, usualmente sin quistes, y en un 60-70 % de los casos localizada en el lóbulo derecho hepático y con un 35 % de pacientes con afectación de ambos lóbulos al diagnóstico. El escáner abdominal y la resonancia magnética confirman el hallazgo, además de permitir establecer la extensión tumoral y su vascularización (Fig. 2).

Figura 2. Hepatoblastoma.
La biopsia del tumor es la única forma de confirmar plenamente el diagnóstico, por lo que se recomienda su realización en todos los casos, y se considera obligada en aquellos que no respondan al perfil clínico habitual (edad menor de 6 meses o mayor de 3 años o cifra de alfa-fetoproteína normal al diagnóstico).
Coexisten dos sistemas de estadiaje, aunque basados en las mismas variables (tamaño y extensión tumoral, posible invasión vascular y existencia de metástasis), para estadiar y estratificar el riesgo de los tumores hepáticos en niños. El propuesto por el Children´s Cancer Study Group (CCSG) y el Children’s Oncology Group (COG) y, desde 1990, un estadiaje adicional (PRETEXT) diseñado por SIOPEL (International Childhood Liver Tumor Strategy Group). Este último es el más utilizado en nuestro entorno europeo y está basado en hallazgos preoperatorios mediante pruebas de imagen, dividiendo los tumores en cuatro categorías (frecuencia relativa de 4 %, 34 %, 29 % y 25 % para estadios I, II, II y IV, respectivamente) según el número de sectores hepáticos (lateral izquierda: segmentos 2 y 3; medial izquierda: segmentos 4a y 4b; anterior derecha: segmentos 5 y 8; y posterior derecha: segmentos 6 y 7) que ocupen, añadiendo a estas una letra según exista o no afectación de vena cava o suprahepáticas (V), vena porta (P), extensión extrahepática abdominal (E), metástasis ganglios linfáticos (N), metástasis a distancia (M) y rotura tumoral/hemorragia intraperitoneal (H)(8) (Fig. 3).
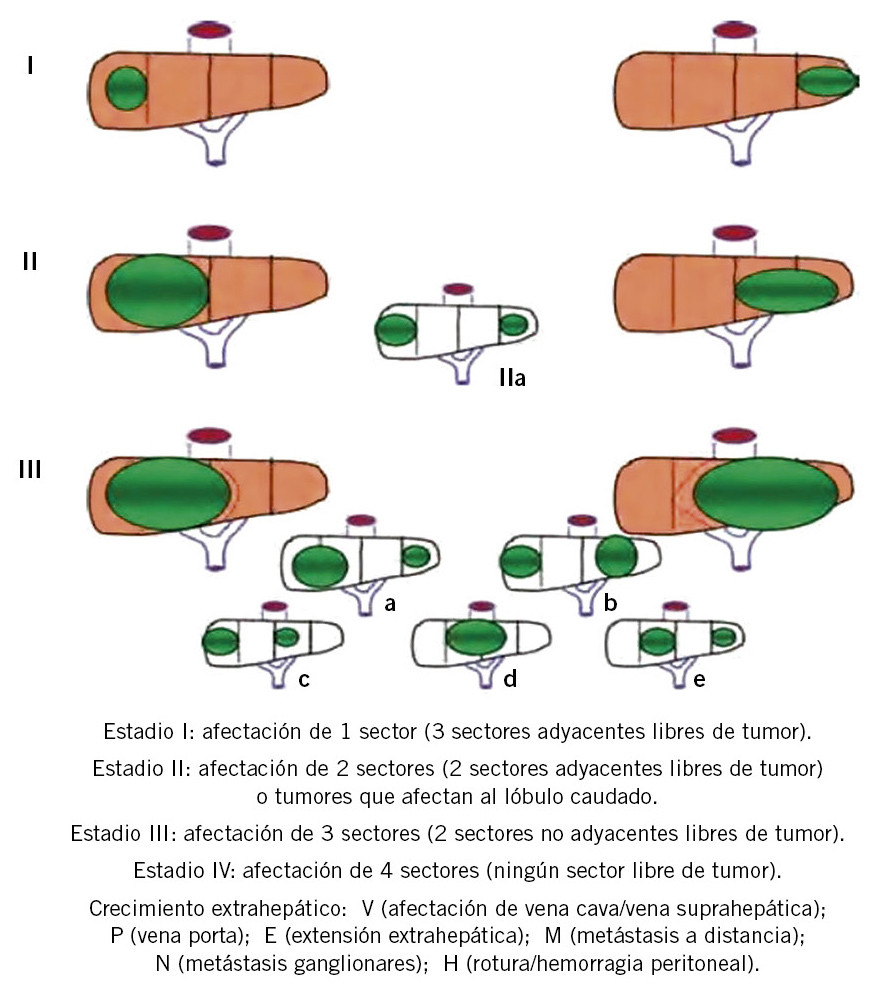
Figura 3. Estadiaje preoperatorio de hepatoblastoma y hepatocarcinoma (PRETEXT).
En cuanto al pronóstico, son criterios de riesgo: el estadio tumoral IV, la presencia de invasión vascular, la extensión ganglionar o metastásica, las formas histológicas más indiferenciadas, la normalidad en los niveles séricos de alfa-proteína y la mayor edad (>8 años) al diagnóstico (Tabla II). La aplicación de estas variables consigue una estratificación del riesgo (muy bajo/bajo/intermedio/alto) de los hepatoblastomas (Tabla II) que permite definir de forma individual el tratamiento más apropiado(9,10).

La posibilidad real de curación del hepatoblastoma se basa en la resección completa del mismo, bien mediante una hepatectomía o mediante un trasplante hepático en aquellos casos considerados como irresecables, y en el tratamiento coadyuvante con quimioterapia(11-13) (Fig. 4).
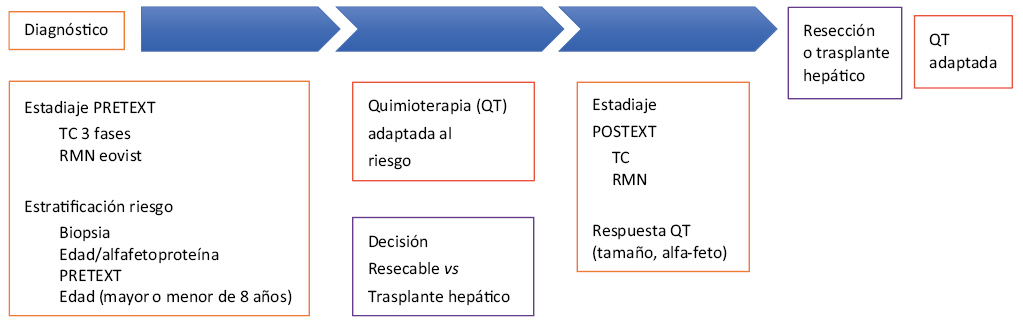
Figura 4. Estrategia de manejo de hepatoblastoma.
El esquema de tratamiento más aceptado recomienda el uso de quimioterapia preoperatoria, porque incrementa la posibilidad de resección completa (desde un 40-60 % hasta un 90 %) al reducir en muchos casos el tamaño tumoral y delimitar mejor su extensión y la postoperatoria.
En referencia a la pauta de quimioterapia, su objetivo es el de lograr la mayor eficacia, pero a costa de la menor toxicidad posible, lo que supone la adaptación de su intensidad (número de ciclos, fármacos incluidos, dosis y frecuencia) basándose en el índice de “riesgo” establecido para cada caso.
En aquellos casos que tras el tratamiento con quimioterapia se mantienen como irresecables (extensión tumoral hepática PRETEXT IV o tumores PRETEXT II-III “centralmente localizados” con afectación de estructuras vasculares hepáticas principales) y no existe evidencia de extensión extrahepática, la única posibilidad de tratamiento es el trasplante hepático(14). La necesidad de trasplante en este tumor se estima entre un 8-10 % y los resultados muestran una tasa de supervivencia global por encima del 80 %, la cual es comparable con la obtenida tras el tratamiento quirúrgico convencional de aquellos tumores considerados como resecables. Los resultados han sido significativamente mejores cuando se ha indicado el trasplante de forma primaria en el momento del diagnóstico frente al trasplante de “rescate” tras una resección tumoral incompleta o recaída de la enfermedad tras dicha intervención (supervivencia libre de enfermedad del 82 % y 30 %, respectivamente). La presencia de metástasis pulmonares en el momento del diagnóstico, si estas desaparecen tras la quimioterapia o son resecadas, no contraindicaría el trasplante, ya que se han obtenido supervivencias de entre el 58 % y 80 % tras el mismo. Estrategias, como la utilización de injertos hepáticos de donante vivo o la priorización de estos pacientes dentro de la lista de espera, reducen el riesgo de recidiva o metástasis postquimioterapia.
La necesidad y el tipo de quimioterapia post-resección o trasplante dependen del nivel de riesgo estimado para cada caso. Su uso ha reducido la tasa de recurrencia de la enfermedad.
A modo de conclusión, el pronóstico de estos tumores viene marcado por la presencia o no de aquellos factores que condicionan su resecabilidad (extensión tumoral, localización extrahepática, invasión vascular, multifocalidad…), estimándose globalmente en un 75 % aproximadamente la posibilidad de curación actual de este tipo de tumor.
Hepatocarcinoma (HCC)
Tumor hepático muy agresivo con una media de 12 años al diagnóstico de los casos sobre hígado sano, aunque también puede desarrollarse sobre cirrosis en niños más pequeños. El pronóstico depende del diagnóstico temprano y de la posibilidad de resección mediante hepatectomía o trasplante hepático.
Tumor de alta malignidad que se desarrolla fundamentalmente en niños mayores (edad media al diagnóstico de 12 años con un 65 % por encima de los diez años de edad) y escasa incidencia (0,24-0,65 caso por millón/año). A diferencia de los adultos, con una práctica totalidad de casos que se desarrollan sobre cirrosis, en la infancia, un 35-40 % de los casos son tumores primarios sobre hígado sano.
En áreas de alta prevalencia de la infección por el virus de la hepatitis B (VHB), la incidencia de HCC secundario ha sido históricamente más alta, pero se ha reducido en un 70-80 % tras la introducción de la vacunación sistemática frente al virus. En nuestro medio, la prevalencia de hepatocarcinoma a largo plazo (10-20 años) en niños con hepatitis crónica por VHB es alrededor del 1 %. Otras enfermedades hepáticas a las que se asocia este tipo de tumor:
• Enfermedad hepática con cirrosis:
– Tirosinemia tipo I.
– Colestasis familiar progresiva: especial riesgo en los tipos 2 (defecto BSEP) y 4 (defecto TJP2).
– Otras: atresia biliar, síndrome de Alagille, déficit de alfa-1 antitripsina. Glucogenosis 3 y 4.
• Enfermedad hepática sin cirrosis:
– Infección crónica por VHB (26-53 % de los HCC son sin cirrosis).
– Glucogenosis tipo 1.
– Shunts portosistémicos.
En la mayoría de tumores primarios, la presentación clínica es en forma de dolor abdominal o detección de una masa abdominal; mientras que en los casos en los que el tumor se desarrolla sobre una cirrosis, son los signos, clínicos o analíticos, derivados de esta los que dominan el cuadro. En dos tercios de los pacientes existe un aumento de alfa-fetoproteína al diagnóstico, y al igual que en el hepatoblastoma, sirve como marcador de respuesta al tratamiento. Las metástasis más frecuentes son pulmonares o de ganglios linfáticos regionales y pueden estar ya presentes en el momento del diagnóstico (31 % y 18 %, respectivamente)(15).
La biopsia hepática es el método de confirmación diagnóstica. Histológicamente, se distinguen dos variantes: (a) el patrón anaplásico o epitelial (75 % de casos), de células más grandes que los hepatocitos normales, con hipercromasia nuclear, dispuestas en trabéculas anchas, pleomorfismo celular con frecuentes células gigantes y ausencia de hematopoyesis; y (b) patrón fibrolamelar (25 %), con células grandes, eosinofílicas en trabéculas separadas por un estroma fibroso lamelar. Esta variante aparece casi invariablemente en adolescentes-adultos jóvenes, sin cirrosis subyacente, con frecuencia no asocia aumento de alfa-fetoproteína y, aunque inicialmente, y en función de la experiencia en pacientes adultos, se consideró como con mejor pronóstico que la variante anaplásica, este hecho no se ha confirmado posteriormente en niños.
El sistema de estadiaje que parece más útil, al igual que sucede con el hepatoblastoma, es el preoperatorio PRETEXT (Fig. 3). La ecografía, el escáner y la resonancia magnética abdominal habitualmente delimitan la masa y pueden definir las posibilidades de resecabilidad (Fig. 5).
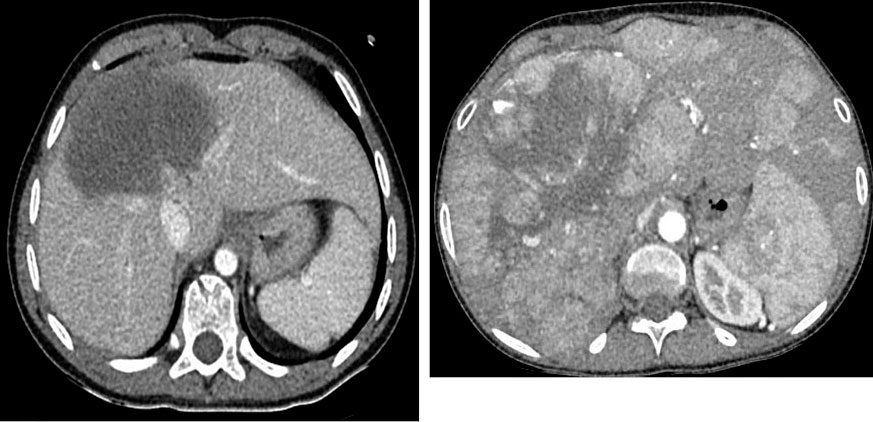
Figura 5. Hepatocarcinoma.
Es habitual que aquellos tumores sobre cirrosis se diagnostiquen en fases precoces (PRETEXT I-II), ya que son pacientes en seguimiento por su enfermedad hepática, mientras que los HCC primarios lo sean en fases tardías (PRETEXT III-IV).
La única opción curativa, al igual que sucede con el hepatoblastoma, se basa en la resección quirúrgica completa del tumor, bien sea mediante realización de hepatectomía en aquellos casos considerados como resecables, o trasplante hepático en el caso de tumores considerados irresecables y siempre con el requisito de ausencia de extensión extrahepática de la enfermedad(16). Además, el trasplante debería ser el tratamiento de elección en todos aquellos HCC que asienten sobre una cirrosis, independientemente de que dicha enfermedad reúna o no criterios de trasplante en el momento del diagnóstico del tumor, ya que se resolverán tanto el tumor como dicha enfermedad subyacente.
Incluso en aquellos casos de tumores primarios considerados como resecables, está actualmente abierta la discusión en cuanto a que el trasplante hepático tenga mejor resultado, en cuanto a supervivencia, frente a la resección mediante hepatectomía parcial.
En pacientes pediátricos, no están claramente definidos los criterios tumorales que contraindiquen el trasplante por alto riesgo de recidiva posterior al mismo, a diferencia de lo que ocurre en pacientes adultos, en los que para la selección de receptores aplican los criterios de Milán (tumores únicos menores de 5 cm o hasta 3 tumores menores de 3 cm o tumor de hasta 7 cm en hígado sano). Datos de registros europeo (ELTR) y norteamericano (UNOS) muestran una supervivencia postrasplante en niños del 53,5 % y 57,6 % a 5 años, respectivamente. Series pequeñas de pacientes han reportado una supervivencia postrasplante a 5 años del 72-83 % en pacientes con HCC que excedían los criterios de Milán.
Se han identificado como factores de riesgo para la recurrencia postrasplante: invasión vascular, tamaño, multifocalidad, trasplante de rescate tras resección fallida y el HCC primario sin enfermedad de base, dado su habitual diagnóstico más tardío.
El resto de opciones disponibles de tratamiento no son curativas. La quimioterapia pre-resección (PLADO: cisplatino, carboplatino y doxorrubicina) obtiene una respuesta parcial en menos del 50 % de casos y no incrementa significativamente la resecabilidad del tumor. Otra opción es el sorafenib, que inhibe el receptor de factor de crecimiento endotelial y Raf kinasa con efecto antiproliferativo/antiangiogénico.
En estos momentos, está activo un estudio (Pediatric Hepatic International Tumor Trial), donde se evalúa la eficacia de quimioterapia asociada a sorafenib pre-resección o trasplante en tumores irresecables o metastásicos y, por otra parte, la quimioterapia post-resección o trasplante en tumores primarios sin enfermedad de base y cuyos resultados no están disponibles actualmente.
El pronóstico del HCC en niños ha mejorado sustancialmente en las dos últimas décadas hasta alcanzar una tasa de supervivencia actual del 60-80 % a 5 años. Los principales factores para esta mejora de los resultados son una detección más precoz y el desarrollo de la técnica quirúrgica y el trasplante hepático, incluyendo el acortamiento de los tiempos de espera en lista, bien mediante la realización de trasplante de donante vivo o split o mediante la priorización en la lista de estos pacientes.
Sarcoma hepático
Tumores muy raros y de alta malignidad con tasas bajas de supervivencia. En algunos casos, los resultados de las pruebas de imagen pueden ser superponibles a un hemangioma, lo que obliga a la biopsia para su diagnóstico diferencial. No existe un tratamiento definido.
Angiosarcoma hepático
Tumor vascular de muy escasa incidencia y alta malignidad. Edad media de presentación entre los tres y cuatro años. La presentación clínica habitual es la de distensión abdominal que puede asociar ictericia, dolor abdominal o disnea. A diferencia de otros tumores vasculares, no suelen asociar un cuadro de insuficiencia cardiaca congestiva. Al diagnóstico, suelen ser tumores grandes con afectación de ambos lóbulos difusamente o con múltiples lesiones. Microscópicamente, es un tumor de células fusiformes de estirpe endotelial. Metastatiza a ganglios linfáticos, suprarrenales, pleura y pulmón. Las pruebas de imagen muestran lesiones hipodensas y heterogéneas, por contener zonas de necrosis y hemorragia, semejantes a las imágenes de un hemangioma hepático infantil. En ocasiones, es difícil el diagnóstico diferencial con este último, y debe sospecharse un angiosarcoma en todo teórico hemangioma que evoluciona progresando de forma inusual o que se diagnostica en un niño mayor de 3-4 años. Su gran malignidad y capacidad de crecimiento determinan un pronóstico muy pobre con una supervivencia global por debajo de los dos años. En su tratamiento se han usado diferentes medidas terapéuticas que incluyen: resección, quimioterapia, radioterapia e incluso trasplante, pero su rareza impide que se haya podido establecer una pauta definida.
Sarcoma embrionario (sarcoma primario)
Es un tumor hepático primitivo, infrecuente, indiferenciado y, generalmente, diagnosticado a partir de los 6-7 años. Algunos estudios sugieren su relación con el hamartoma mesenquimal, bien como polos opuestos en un mismo espectro o bien como un estadio evolutivo posterior de este. Macroscópicamente, aparece como un tumor grande, sólido pero con áreas quísticas. Tradicionalmente, considerado de muy mal pronóstico, en años recientes la combinación en su tratamiento de quimioterapia o radioterapia previa y resección quirúrgica posterior ha permitido alcanzar tasas de supervivencia de hasta un 80 % a los 4 años.
Rabdomiosarcoma de la vía biliar
Tumor extremadamente raro que aparece casi exclusivamente durante la infancia (con frecuencia, en menores de 5 años). Debut clínico con ictericia, hepatomegalia, coluria e hipocolia. El diagnóstico se hace por biopsia, en algunos casos a través de una colangiografía endoscópica retrógrada o transparietohepática. Pobre supervivencia en general, aunque se han reportado casos con buena evolución tras quimioterapia y radioterapia y subsiguiente resección y quimioterapia posterior a esta.
Función del pediatra de Atención Primaria
En la mayoría de los casos, los tumores hepáticos son asintomáticos o presentan síntomas inespecíficos, siendo la palpación de una hepatomegalia el primer paso para su diagnóstico. Por este motivo, es en el ámbito de la Atención Primaria donde se establece la sospecha por parte del pediatra en las revisiones de rutina del niño sano o ante la consulta por dolor abdominal o molestias digestivas. En el caso de la detección de una hepatomegalia dura, es necesaria la remisión urgente del paciente para la realización de una primera prueba de imagen (ecografía) que establezca el diagnóstico de un tumor hepático. La tipificación histopatológica, la definición del tratamiento y el mismo se realizan en el ámbito hospitalario; mientras que, en el seguimiento posterior, la intervención combinada de especialista (hepatólogo u oncólogo pediátrico) y pediatra de Atención Primaria, este último en los aspectos de seguimiento pediátrico (nutrición, vacunas, desarrollo psicomotor, etc.), diagnóstico y tratamiento de enfermedades intercurrentes (infecciones) y detección de posibles complicaciones o efectos secundarios del tratamiento de la enfermedad tumoral. En referencia a los hemangiomas es importante ante la detección de angiomas cutáneos grandes o muy numerosos la búsqueda de lesiones viscerales / hepáticas mediante prueba de imagen (ecografía).
Conflicto de intereses
No hay conflicto de interés en la elaboración del presente manuscrito ni fuente de financiación.
Bibliografía
Los asteriscos muestran el interés del artículo a juicio de los autores.
1. Meyers RL. Tumors of the liver in children. Surg Oncol. 2007; 16: 195-203.
2. Meyers R, Hiyama E, Czauderna P, Tiao GM. Liver Tumors in Pediatric Patients. Surg Oncol Clin N Am. 2021; 30: 253-74.
3. Christison-Lagay ER, Burrows PE, Alomari A, Dubois J, Kozakewich HP, Lane TS, et al. Hepatic hemangiomas: subtype classification and development of a clinical practice algorithm and registry. J Pediatr Surg. 2007; 42: 62-7.
4. Iacobas I, Phung TL, Adams DM, Trenor CC 3rd, Blei F, Fishman DS, Hammill A, et al. Guidance Document for Hepatic Hemangioma (Infantile and Congenital) Evaluation and Monitoring. J Pediatr. 2018; 203: 294-300.e2.
5. Krowchuk DP, Frieden IJ, Mancini AJ, Darrow DH, Blei F, Greene AK, et al; Subcommittee on the Management of Infantile Hemangiomas. Clinical Practice Guideline for the Management of Infantile Hemangiomas. Pediatrics. 2019; 143: e20183475. Disponible en: https://doi.org/10.1542/peds.2018-3475.
6. Mazereeuw-Hautier J, Hoeger PH, Benlahrech S, Ammour A, Broue P, Vial J, et al. Efficacy of propranolol in hepatic infantile hemangiomas with diffuse neonatal hemangiomatosis. J Pediatr. 2010; 157: 340-2.
7. Léauté-Labrèze C, Hoeger P, Mazereeuw-Hautier J, Guibaud L, Baselga E, Posiunas G, et al. A randomized, controlled trial of oral propranolol in infantile hemangioma. N Engl J Med. 2015; 372: 735-46.
8. Roebuck DJ, Aronson D, Clapuyt P, Czauderna P, de Ville de Goyet J, Gauthier F, et al; International Childhood Liver Tumor Strategy Group. 2005 PRETEXT: a revised staging system for primary malignant liver tumours of childhood developed by the SIOPEL group. Pediatr Radiol. 2007; 37: 123-32.
9. Meyers RL, Rowland JR, Krailo M, Chen Z, Katzenstein HM, Malogolowkin MH. Predictive power of pretreatment prognostic factors in children with hepatoblastoma: a report from the Children’s Oncology Group. Pediatr Blood Cancer. 2009; 53: 1016-22.
10. Aronson DC, Schnater JM, Staalman CR, Weverling GJ, Plaschkes J, Perilongo G, et al. Predictive value of the pretreatment extent of disease system in hepatoblastoma: results from the International Society of Pediatric Oncology Liver Tumor Study Group SIOPEL-1 study. J Clin Oncol. 2005; 23: 1245-52.
11. Perilongo G, Shafford E, Maibach R, Aronson D, Brugieres L, Brock P, et al; International Society of Paediatric Oncology-SIOPEL 2. Risk-adapted treatment for childhood hepatoblastoma. final report of the second study of the International Society of Paediatric Oncology–SIOPEL 2. Eur J Cancer. 2004; 40: 411-21.
12. Zsíros J, Maibach R, Shafford E, Brugieres L, Brock P, Czauderna P, et al. Successful treatment of childhood high-risk hepatoblastoma with dose-intensive multiagent chemotherapy and surgery: final results of the SIOPEL-3HR study. J Clin Oncol. 2010; 28: 2584-90.
13. Otte JB. Progress in the surgical treatment of malignant liver tumors in children. Cancer Treat Rev. 2010; 36: 360-71.
14. Otte JB, de Ville de Goyet J, Reding R. Liver transplantation for hepatoblastoma: indications and contraindications in the modern era. Pediatr Transplant. 2005; 9: 557-65.
15. Czauderna P, Mackinlay G, Perilongo G, Brown J, Shafford E, Aronson D, et al; Liver Tumors Study Group of the International Society of Pediatric Oncology. Hepatocellular carcinoma in children: results of the first prospective study of the International Society of Pediatric Oncology group. J Clin Oncol. 2002; 20: 2798-804.
16. McAteer JP, Goldin AB, Healey PJ, Gow KW. Surgical treatment of primary liver tumors in children: outcomes analysis of resection and transplantation in the SEER database. Pediatr Transplant. 2013; 17: 744-50. Disponible en: https://doi.org/10.1111/petr.12144.
Bibliografía recomendada
– Meyers R, Hiyama E, Czauderna P, Tiao GM. Liver Tumors in Pediatric Patients. Surg Oncol Clin N Am. 2021; 30: 253-74.
Revisión actualizada y amplia de los diferentes tumores hepáticos en la infancia. Extensa bibliografía sobre el tema.
– Krowchuk DP, Frieden IJ, Mancini AJ, Darrow DH, Blei F, Greene AK, et al; Subcommittee on the Management of Infantile Hemangiomas. Clinical Practice Guideline for the Management of Infantile Hemangiomas. Pediatrics. 2019; 143: e20183475. Disponible en: https://doi.org/10.1542/peds.2018-3475.
Primera guía de práctica clínica de la American Academy of Pediatrics (AAP) sobre el manejo de los hemangiomas infantiles en su localización cutánea o visceral.
– McAteer JP, Goldin AB, Healey PJ, Gow KW. Surgical treatment of primary liver tumors in children: outcomes analysis of resection and transplantation in the SEER database. Pediatr Transplant. 2013; 17: 744-50. Disponible en: https://doi.org/10.1111/petr.12144.
Análisis riguroso de los resultados de la hepatectomía versus el trasplante hepático en el número importante de tumores incluidos en el registro de cáncer SEER (The Surveillance, Epidemiology, and End Results Program).
– Czauderna P, Mackinlay G, Perilongo G, Brown J, Shafford E, Aronson D, et al; Liver Tumors Study Group of the International Society of Pediatric Oncology. Hepatocellular carcinoma in children: results of the first prospective study of the International Society of Pediatric Oncology group. J Clin Oncol. 2002; 20: 2798-804.
Estudio prospectivo sobre hepatocarcinoma del grupo de expertos en el contexto de la Sociedad Internacional de Oncología Pediátrica.
| Caso clínico |
|
Niño de 5 meses que consulta por episodios de llanto y haber notado los padres que el abdomen en su zona superior derecha está duro. No presenta otra sintomatología. Antecedentes perinatales sin interés. Curva de peso y talla normal. Sin antecedentes familiares de interés. Exploración física: buen estado general. Bien nutrido. Palidez cutánea. Auscultación cardíaca sin soplos. Auscultación pulmonar: buena ventilación bilateral. Abdomen blando y depresible. No doloroso. Se palpa una hepatomegalia de tres traveses de dedo, de consistencia dura y borde irregular. No se palpa bazo. Sin adenopatías. Neurológico normal.
|

 Diagnosis in liver disease
Diagnosis in liver disease