 |
| Temas de FC |
M.J. Quiles Blanco![]() , L. Fernández Tomé
, L. Fernández Tomé
Facultativo Especialista Adjunto. Servicio de Hepatología y Trasplante Hepático Infantil. Hospital Universitario La Paz. Madrid
| Resumen
Las enfermedades hepáticas en la infancia abarcan una amplia gama de patologías, que incluyen: colestasis, enfermedades metabólicas, autoinmunes, fibroquísticas, tumores e infecciones. El diagnóstico se basa en una anamnesis detallada y pruebas complementarias que permiten identificar la enfermedad, evaluar su pronóstico y respuesta al tratamiento. Las pruebas de laboratorio son fundamentales para detectar daño hepático, insuficiencia hepática o estudiar un paciente con colestasis. Los marcadores utilizados principalmente incluyen transaminasas AST (aspartato aminotransferasa), ALT (alanina aminotransferasa), bilirrubina, GGT (gamma-glutamil transferasa) y la coagulación como marcador de función hepática. Además, las técnicas de imagen y los estudios genéticos complementan el diagnóstico, permitiendo un manejo integral. El pediatra en Atención Primaria juega un papel crucial en la detección temprana y seguimiento de estas patologías, como en la ictericia prolongada o hipertransaminasemia persistente. |
| Abstract
Liver diseases in childhood encompass a wide range of conditions, including cholestasis, metabolic, autoimmune, fibro-cystic diseases, tumors, and infections. Diagnosis begins with a thorough medical history and complementary tests, allowing for disease identification, prognosis evaluation, and treatment response monitoring. Laboratory tests are essential to detect liver damage, liver failure, or cholestasis. Key markers include transaminases AST (aspartate aminotransferase), ALT (alanine aminotransferase), bilirubin, GGT (gamma-glutamyl transferase) and clotting. Additionally, imaging techniques and genetic studies complement the diagnostic approach, enabling comprehensive management. Primary care pediatricians play a vital role in the early detection and follow-up of these conditions, such as prolonged jaundice or persistent hypertransaminasemia. |
Palabras clave: Enfermedad hepática; Colestasis; Hipertensión portal; Daño hepático; Infecciones.
Key words: Liver disease; Cholestasis; Portal hypertension; Liver damage; Infections.
Pediatr Integral 2025; XXIX (1): 16 – 24
OBJETIVOS
• Identificar y evaluar signos clínicos en la enfermedad hepática en el niño y realizar una anamnesis dirigida. Además, aprender qué pruebas solicitar según la sospecha.
• Interpretar resultados de pruebas de laboratorio para enfocar el diagnóstico de un niño con sospecha de hepatopatía.
• Establecer criterios de gravedad que impliquen referencia a un centro especializado, principalmente conocer e identificar los signos de alarma (encefalopatía, coagulopatía, hipertensión portal).
 |
 |
Diagnóstico en la enfermedad hepática
https://doi.org/10.63149/j.pedint.3
Introducción
Las enfermedades hepáticas en la infancia son muy diversas. Las podemos dividir en grandes grupos: colestasis, enfermedades metabólicas, enfermedades autoinmunes, enfermedades fibroquísticas, tumores o infecciones(1). El diagnóstico siempre comienza con la historia clínica, añadiendo después las pruebas complementarias pertinentes. Estos test diagnósticos nos permiten realizar un despistaje de la enfermedad hepática, evaluar su pronóstico y su respuesta a tratamiento una vez se ha instaurado.
Historia clínica
La anamnesis completa y detallada es esencial para orientar el diagnóstico en pacientes con sospecha de enfermedad hepática, incluyendo antecedentes personales, familiares y gestacionales. La exploración física busca signos clínicos, como hepatoesplenomegalia o estigmas de hepatopatía.
La anamnesis detallada será fundamental para orientar las pruebas necesarias en un paciente con sospecha de hepatopatía: historia clínica completa con antecedentes personales y familiares, historia gestacional, ecografías y analíticas prenatales, tipo de parto, necesidad de reanimación, peso al nacer, cribado metabólico, auditivo, curva pondoestatural, aversiones en la diversificación alimentaria y desarrollo psicomotor. Además, es importante conocer si existe antecedente de tratamiento farmacológico o productos de herbolario u homeopatía.
En cuanto a antecedentes familiares, se valorará la presencia de autoinmunidad en la familia, antecedentes obstétricos (prurito gestacional), antecedentes de malformaciones o muertes fetales, consanguinidad, cálculos o quistes en la familia, entre otros. Se deben orientar las preguntas según la sospecha.
Sobre la exploración física, destacar que en la hepatopatía crónica los signos clínicos aparecen tarde y no son específicos de una enfermedad concreta. La ictericia se observa en la hiperbilirrubinemia; a partir de 3 mg/dL se hace evidente en escleras. Las causas son diversas y no siempre tienen que ver con enfermedad hepática (Tabla I).

La eritrosis palmar o palmas hepáticas es un eritema punteado en las yemas de los dedos y en la región tenar e hipotenar por vasodilatación (Fig. 1).
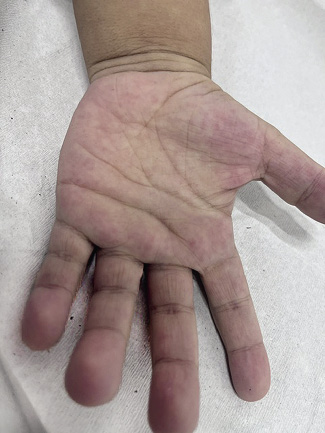
Figura 1. La eritrosis palmar o palmas hepáticas es un eritema punteado en las yemas de los dedos y en las regiones tenar e hipotenar por vasodilatación.
Las arañas vasculares pueden localizarse en cara, abdomen o dorso de las manos (Fig. 2).

Figura 2. Spider. Las arañas vasculares pueden localizarse en cara, abdomen o dorso de las manos.
Datos de desarrollo de hipertensión portal, como complicación de la enfermedad, son: la detección de circulación colateral (vasos distendidos y marcados en hipocondrio y abdomen) y hepato o esplenomegalia (Fig. 3).

Figura 3. Ascitis e ictericia junto con otros datos de hepatopatía crónica grave, como circulación colateral en paciente con atresia de vía biliar sin restablecimiento de flujo tras Kasai.
Algunos pacientes, en la evolución de una hepatopatía avanzada, pueden presentar ascitis y acropaquias por la existencia de shunts arteriovenosos intrapulmonares. Los xantomas son depósitos de lípidos en cara y zonas de extensión, se ven en colestasis graves junto con lesiones de rascado por prurito(2). Algunas enfermedades pueden presentar un fenotipo concreto (síndrome de Alagille, infecciones TORCH, hipopituitarismo, síndrome de Zellweger, trastornos de glicosilación) o presencia de soplo cardiaco. El 10 % de los pacientes con atresia de vía biliar pueden asociar situs inversus (formas “sindrómicas”), pudiendo variar la auscultación. La acantosis nigricans es un hallazgo frecuente en niños con enfermedad de hígado graso no alcohólico (EHGNA). La encefalopatía hepática es difícil de valorar, sobre todo en niños de menor edad; inicialmente, consiste en cambios sutiles en el comportamiento, pero a medida que avanza será más evidente la somnolencia y, en estadios finales, coma arreactivo. Se da en situaciones de fallo hepático agudo en niños previamente sanos o en hepatópatas crónicos que sufren una descompensación, como puede ser un episodio de sangrado digestivo o infección.
La hepatomegalia puede estar presente en enfermedades hepáticas y extrahepáticas. Los mecanismos etiopatogénicos implicados son: inflamación (infección, autoinmunidad), depósito (glucogenosis, obesidad), infiltración, congestión vascular y la obstrucción biliar.
La esplenomegalia se detecta en enfermedades que han desarrollado hipertensión portal, como hepatitis autoinmune, obstrucción extrahepática al flujo portal (cavernoma) o en enfermedades de depósito (glucogenosis y enfermedades lisosomales).
Laboratorio
El hígado realiza funciones clave en síntesis, almacenamiento y depuración, y su evaluación frecuentemente comienza con análisis que pueden ser inespecíficos. En Pediatría, es importante interpretar los valores en función de la edad.
El hígado es un órgano envuelto en importantes funciones en el organismo: síntesis, almacenamiento, excreción y depuración de numerosas sustancias. El diagnóstico de las enfermedades hepáticas comienza muchas veces en una extracción analítica donde poder valorar todas ellas. Cualquier parámetro analítico alterado debe ser analizado en el contexto concreto del paciente. En general, son inespecíficos y no determinan el tipo de hepatopatía, incluso un paciente con hepatopatía crónica se puede presentar con transaminasas normales. Otra posibilidad es la detección de una enfermedad sistémica sin implicación del hígado, aunque presente alteración en bioquímica hepática, como ocurre en situaciones de shock o hipoperfusión sistémica o en las infecciones respiratorias tan frecuentes en la infancia.
Es fundamental en Pediatría disponer de correctas referencias por edad para todos los parámetros analizados, por ejemplo: un neonato puede tener hasta cinco veces elevada la GGT sobre el límite del valor normal(3).
Clásicamente, los parámetros que pueden ser analizados se clasifican según la información que dan sobre cómo está el hígado: si existe daño hepático, si hay insuficiencia hepática o si hay colestasis.
Daño hepático
Las transaminasas AST y ALT son enzimas hepáticas clave para evaluar el daño en el hígado; AST también se encuentra en otros tejidos, por lo que su elevación no es específica de daño hepático. ALT es más específica del hígado, y el análisis conjunto de ambas ayuda a identificar la causa y severidad del daño.
Transaminasas
Las transaminasas son enzimas intracelulares que catalizan la transferencia del grupo alfa-amino (NH2) de aminoácidos aminados (AST: ácido aspártico, ALT: alanina) al grupo alfa-ceto del ácido cetoglutárico.
AST se encuentra en más células además de en el hígado: músculo, corazón, riñón, páncreas, cerebro, leucocitos y eritrocitos. La elevación de AST, por tanto, no es específica y en el diagnóstico diferencial se debe solicitar creatinfosfoquinasa (CPK) para discriminar el origen muscular de la elevación de AST (si está elevada la CPK, sospecharemos esfuerzo físico excesivo o distrofia muscular, entre otros), o haptoglobina y LDH (lactato deshidrogenasa) por si es de origen eritrocitario.
La enzima ALT es más específica, se encuentra en el citosol del hepatocito. Ambas se deben estudiar de forma conjunta, ya que la proporción entre ambas puede orientar sobre la causa subyacente. AST está presente, además de en el citosol, en la mitocondria del hepatocito; su elevación puede implicar necrosis más grave. Una relación de transaminasas “invertidas” (AST mayor que ALT) indica un daño hepático más profundo.
Existen tablas con percentiles y variabilidad por sexo, pero en la práctica clínica se suele usar un punto de corte de 45 UI/L. No obstante, no hay una relación directa entre la cifra de transaminasas y la gravedad de la enfermedad subyacente. Las transaminasas se liberan a sangre tras el daño en la membrana del hepatocito y hay una mala relación entre el grado de necrosis en biopsias hepáticas y la elevación de transaminasas obtenidas.
El diagnóstico diferencial de una hipertransaminasemia asintomática incluirá: enfermedad hepática grasa no alcohólica (EHGNA), enfermedad de Wilson, déficit de alfa-1 antitripsina, hepatitis virales, tóxicos, enfermedad autoinmune y fructosemia (Tabla II).

La enfermedad del hígado graso no alcohólico es la causa más frecuente de enfermedad hepática en niños en EE.UU., dado el auge de la obesidad infantil: es importante su diagnóstico y seguimiento precoz por su alta prevalencia y su buena evolución con tratamiento adecuado. La asociación con síndrome metabólico, cirrosis y hepatocarcinoma lo ha hecho una causa mayoritaria de indicación de trasplante en adultos(4,5) (Tabla III).

Ante cualquier hipertransaminasemia, es importante realizar un seguimiento hasta asegurar la resolución analítica o el diagnóstico del paciente, aunque la mayoría de los casos tengan resolución espontánea, no anula la necesidad de estudio y seguimiento.
Los signos de gravedad asociados son: los valores más altos (diez veces el valor normal), asociar alteración de GGT y/o bilirrubina, presentar datos de insuficiencia hepática (coagulopatía o sangrados como epistaxis) o signos de hipertensión portal (esplenomegalia o trombopenia)(6).
LDH
La enzima lactato deshidrogenasa (LDH) está presente en músculo cardiaco y esquelético, eritrocitos, riñón y cerebro. Se eleva en isquemia y hemólisis. En el caso de una elevación mantenida de LDH y fosfatasa alcalina, hay que descartar infiltración maligna del hígado.
Función de síntesis
La albúmina, principal proteína sanguínea sintetizada en el hígado, y los factores de coagulación son indicadores clave de la función hepática. Las lipoproteínas y la colinesterasa también ayudan a evaluar la gravedad y evolución de las hepatopatías.
Albúmina
La albúmina es la principal proteína de la sangre, está sintetizada únicamente en el hígado y tiene una vida media de 20 días; por tanto, niveles bajos suelen ser indicativos de patología crónica. El diagnóstico diferencial de la hipoalbuminemia incluirá la enteropatía pierde-proteínas, el síndrome nefrótico o la desnutrición grave. Sus funciones no solo incluyen regular la presión oncótica, tiene efecto inmunomodulador, antioxidante, estabiliza el endotelio y transporta diversas moléculas (incluyendo fármacos y toxinas).
Coagulación
El hígado sintetiza muchas proteínas de la cascada de la coagulación (procoagulantes y anticoagulantes): factores V, VII, IX, X, XI, protrombina y fibrinógeno. La vitamina K es necesaria en varias de las reacciones y la capacidad de almacenamiento del hígado es limitada; por ello, podemos ver en situaciones de déficit (colestasis o diarrea) que se alargue el tiempo de protrombina y el INR (índice internacional normalizado), pero se resolverá tras la administración intramuscular o intravenosa de vitamina K.
El fallo hepático agudo se define por un INR elevado (o tiempo de protrombina alargado y actividad de protrombina baja), pero hay un equilibrio entre los factores pro y anticoagulantes y, por tanto, no refleja el riesgo de sangrado ni la necesidad de recibir transfusión de hemoderivados. Es una situación crítica que necesita manejo en unidades de trasplante hepático infantil. Ante un valor de actividad de protrombina disminuido o INR aumentado, se debe administrar vitamina K para valorar la reversibilidad del cuadro y, si no es así, referir a un centro especializado(7,8).
El fibrinógeno bajo se puede ver en cuadros de coagulación intravascular diseminada o en hepatopatía avanzada. Existen enfermedades genéticas con mutaciones específicas que afectan a su síntesis: afibrinogenemia (herencia autosómica recesiva), disfibrinogenemia o hipofibrinogenemia (herencia auosómica dominante). El fibrinógeno puede elevarse como reactante de fase aguda.
El factor VIII no se sintetiza en el hígado, por tanto, sirve para diferenciar alteración en la coagulación debida a hepatopatía (factor VIII normal) de la debida a coagulación intravascular diseminada (factor VIII disminuido).
Liproproteínas
El hígado sintetiza y metaboliza muchas de las lipoproteínas. En la colestasis hay un aumento en la síntesis de colesterol inducido por la regurgitación de fosfolípidos a la circulación plasmática. Además, se forma lipoproteína X, que es una forma anómala de LDL (lipoproteína de baja densidad).
En la hepatopatía no colestásica, la bajada en colesterol y lipoproteínas indica empeoramiento de la disfunción y peor pronóstico.
En el daño hepático agudo hay hipertrigliceridemia y aumento de LDL por descenso de la actividad de LCAT (lecitin colesterol acetil transferasa) y de la lipasa de triglicéridos.
Por otra parte, la alteración en las cifras de las lipoproteínas es inespecífica y no permite discernir la causa de la hepatopatía. En las patologías que elevan mucho la cifra de colesterol aparecen xantomas, como en el síndrome de Alagille.
Colinesterasa
La colinesterasa (CHE) cataliza la hidrólisis de los ésteres de colina. Se sintetiza en hígado, páncreas e intestino delgado y, por ello, es indicativo de hepatopatía o de progresión de la misma, porque indica incapacidad en la síntesis de proteínas. Hay gran variabilidad interindividual, pero es útil para ver la progresión en analíticas sucesivas en el mismo paciente.
Puede estar disminuida también en desnutrición, insuficiencia cardiaca congestiva y en el déficit congénito de colinesterasa (se produce en el 4 % de la población).
Función detoxificadora
El hígado detoxifica el amonio mediante el ciclo de la urea. En casos de daño hepático, el amonio se acumula en sangre y afecta al cerebro, provocando encefalopatía en fallos hepáticos agudos o cirrosis descompensada.
Amonio
El amonio se sintetiza en el colon por la ureasa de las bacterias intestinales que degradan las proteínas de la dieta y su metabolización se produce en el hígado mediante el ciclo de la urea, convirtiéndose en urea y glutamina.
El daño hepático implica la liberación de amonio a sangre, produciendo un efecto deletéreo sobre el tejido cerebral. La encefalopatía hepática en niños tiene mala correlación con la cifra de amonio. Puede producirse en casos de fallo hepático agudo o en episodios de descompensación sobre cirrosis; por ejemplo, en un episodio de sangrado intestinal (descompensación aguda sobre enfermedad crónica: acute-on-chronic).
Otra causa de hiperamonemia es la presencia de shunts portosistémicos que pueden ser congénitos o aparecer en un hígado cirrótico con obstrucción a la entrada de sangre por la vena porta o en la enfermedad portosinusoidal.
Además, parte del amonio se sintetiza en el intestino delgado y el riñón, y puede ser inducido por ciertos fármacos, como el ácido valproico.
Daño en el flujo biliar
La colestasis es una disminución en el flujo biliar que acumula bilirrubina y otros compuestos en sangre y su diagnóstico implica evaluar bilirrubina, fosfatasa alcalina (FA), GGT y ácidos biliares, entre otros. La bilirrubina conjugada elevada siempre indica daño hepático.
Se denomina colestasis a la situación en la que el flujo biliar está disminuido, dando lugar al acúmulo en plasma de ácidos biliares, colesterol y bilirrubina. En la biopsia hepática, se define por la presencia de pigmento biliar en los hepatocitos y conductos biliares(9).
La colestasis está asociada con alteraciones complejas transcripcionales en los transportadores de sales biliares y enzimas que participan en la síntesis biliar. Es un campo de investigación en crecimiento, en el que se están describiendo nuevos genes implicados que ayudan a comprender el mecanismo molecular de las colestasis intrahepáticas familiares y a ofrecer nuevas dianas terapéuticas.
Bilirrubina
La bilirrubina es un pigmento derivado de la metabolización del grupo hemo de la hemoglobina de los eritrocitos destruidos en bazo, médula ósea e hígado, y una pequeña fracción tiene su origen en la destrucción de proteínas que contienen hemo: mioglobina, citocromos y peroxidasas. La bilirrubina no conjugada será introducida en el hepatocito, donde será conjugada con la enzima UDP (uridina difosfato) glucuroniltransferasa, siendo soluble y pudiendo ser excretada a la bilis por el canalículo biliar para posteriormente dar color a las heces.
La cifra de bilirrubina se verá alterada en patologías con colestasis o en aquellas patologías con hígado insuficiente, en el cual el metabolismo de la misma estará también afectado.
La bilirrubina conjugada se une a albúmina y tiene mayor vida media, hasta 14 días; esto explica que, en procesos obstructivos reversibles, persista elevada una vez se han resuelto los mismos.
La hiperbilirrubinemia indirecta o no conjugada (liposoluble) se puede detectar en: hemólisis (ver LDH); síndrome de Crigler-Najjar (déficit congénito de UDP glucuroniltransferasa); síndrome de Gilbert (presente en el 5 % de la población sana, hiperbilirrubinemia en contexto de cuadro infeccioso o de estrés sin implicar hepatopatía crónica); o en la hiperbilirrubinemia fisiológica del recién nacido, que tiene un origen multifactorial en relación con la inmadurez hepática neonatal(10). Cuando se sobrepasa la capacidad de transporte unida a la albúmina, la fracción libre atraviesa la barrera hematoencefálica, produciendo lesión cerebral (kernicterus).
La hiperbilirrubinemia conjugada se define por un valor de bilirrubina directa más del 20 % de la bilirrubina total o de más de 1 mg/dL si la bilirrubina total es <5 mg/dL. Indica siempre daño hepatobiliar y es siempre patológica. Se puede detectar en orina con tira reactiva de forma rápida y barata, puede ayudar, pero no se suele utilizar, porque factores externos, como pH o alteración tubular, pueden modificarlo. No se deben demorar los estudios en estos pacientes, ya que puede tener implicaciones pronósticas; por ejemplo, la detección y tratamiento precoz en la galactosemia, panhipopituitarismo o sepsis, en el caso de un neonato con colestasis antes de que aparezcan secuelas, o de una atresia de vía biliar o quiste de colédoco antes de que el daño sea irreversible. La cifra de bilirrubina no diferencia si la enfermedad es intra o extrahepática(11) (Tabla IV).

Fosfatasa alcalina
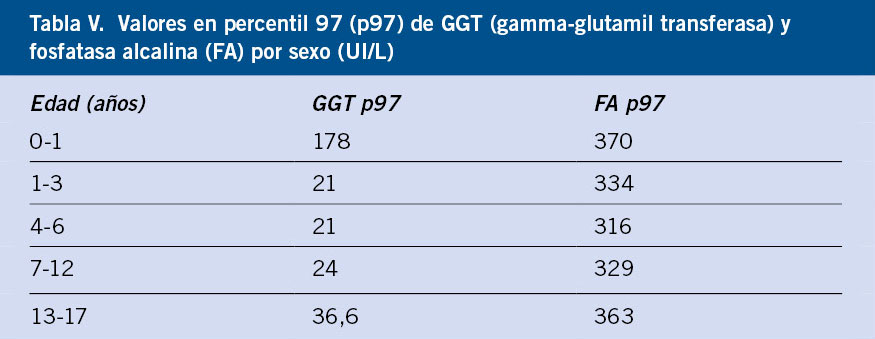
La fosfatasa alcalina (FA) es un grupo de enzimas implicadas en metabolizar ésteres de fosfato a fosfato inorgánico, se encuentra en numerosas células: membrana canalicular del hepatocito, osteoblasto, borde en cepillo del enterocito, túbulo proximal renal, leucocitos y placenta. No está clara su función a nivel hepático. Una elevación aislada en FA en un niño en crecimiento (precisamente por su implicación en la síntesis de hueso) sin otros test hepáticos alterados no indica hepatopatía, existen tablas de percentiles con los valores normales por edad de FA(12) (Tabla V).
Existe una entidad llamada hiperfosfatasemia benigna de la infancia en la que permanece alterada semanas sin otra causa aparente (hasta 10 veces el valor normal), también puede verse elevada en diarreas. Habrá que hacer un control analítico posterior para comprobar normalidad y descartar otros datos que impliquen diferentes patologías (LDH en tumores, GGT en procesos obstructivos biliares).
GGT
La gamma glutamiltransferasa es una enzima que cataliza la transferencia de grupos glutamilo de péptidos, como glutatión, a otros aminoácidos. También está presente en muchas células: riñón, páncreas, hígado, bazo, cerebro e intestino delgado; por tanto, de forma aislada es inespecífica. Ayuda a discriminar el origen hepático de una FA elevada. Los neonatos tienen valores más elevados, normalizándose alrededor de los 6-9 meses de edad (Tabla V).
Es una enzima inducible por fármacos y también estará elevada en procesos obstructivos de la vía biliar: atresia de vía biliar, colangitis esclerosante y síndrome de Alagille, entre otros. No diferencia entre procesos obstructivos intra o extrahepáticos. En las colestasis intrahepáticas familiares, se emplea como clasificación tener GGT elevada o normal. Existe una entidad llamada colestasis benigna recurrente (autosómica recesiva) que tiene un carácter intermitente, pudiendo durar el episodio de colestasis y prurito semanas o meses. El locus es el mismo que el de la colestasis intrahepática familiar por defecto de FIC1. A veces, se desencadena con el embarazo o con el empleo de anticonceptivos orales (Tabla VI).

Ácidos biliares
Los ácidos biliares se sintetizan en el hepatocito y son conjugados con glicina o taurina y excretados a la bilis para participar en la digestión de la grasa de la dieta. Además, activan receptores nucleares que participan en diversas funciones: controlan la homeostasis de la síntesis de los propios ácidos biliares y en el metabolismo de fármacos, lípidos y glucosa. Es un campo en expansión como diana farmacológica para varias enfermedades.
En la hepatopatía se altera el metabolismo de ácidos biliares por destrucción de la masa celular, disminución de su excreción y la aparición de shunts portosistémicos. El defecto congénito de la síntesis de ácidos biliares es una enfermedad con baja prevalencia que se diagnostica midiendo el nivel de los mismos en orina. No puede medirse si el paciente recibe ácido ursodeoxicólico.
Miscelánea
Globulinas séricas
Las globulinas séricas se clasifican según su migración en electroforesis:
• Alfa-1 que incluye, principalmente, alfa-1 antitripsina, ceruloplasmina y orosomucoide, todas actúan como reactantes de fase aguda.
En el déficit de alfa-1 antitripsina, además de la patología pulmonar en el adulto (enfisema precoz), en el niño se desarrolla hepatopatía por acúmulo de proteína mal plegada. Se relaciona con el genotipo, de forma que no todas las mutaciones producen enfermedad. La ceruloplasmina participa en el transporte de cobre en sangre y está baja en la enfermedad de Wilson.
• Alfa-2: incluye haptoglobina, también puede comportarse como reactante de fase aguda.
• Beta: las principales son transferrina y betalipoproteína.
• Gamma: las gammaglobulinas son IgG, IgM e IgA. En la hepatitis autoinmune hay hipergammaglobulinemia a expensas de IgG, están sintetizadas por linfocitos B, por lo que no son exactamente un indicador de función hepática.
Aminoácidos
La medición de aminoácidos en sangre y orina se emplea para el diagnóstico de defectos congénitos del metabolismo, como los defectos del ciclo de la urea, la tirosinemia o las acidemias orgánicas (metilmalónica, propiónica).
Autoanticuerpos
Aunque no se sintetizan en el hígado, en el diagnóstico diferencial de la hepatopatía es frecuente hacer el despistaje de enfermedades autoinmunes: antitransglutaminasa IgA, anti-LKM (anticuerpos de tipo 1 microsomales de hígado y riñón), anticitosol hepático (LC1), antinuclear (ANA) y antimúsculo liso(13).
Alfa-fetoproteína
Marcador tumoral, sobre todo con especial relevancia en el hepatocarcinoma, aunque no siempre se eleva. Puede servir, además de para diagnóstico, para evaluar recidiva tras tratamiento.
Microbiología
El diagnóstico en la enfermedad hepática, en muchas ocasiones, va orientado a la infección viral en el niño sano o hijo de madre con infección por virus de la hepatitis B o C derivado desde maternidad. Por tanto, en la batería de pruebas diagnósticas se incluyen: serologías TORCH, citomegalovirus, virus Epstein Barr (VEB), hepatitis A, B, C y E. Las hepatitis agudas virales asocian mucha citolisis, pueden ser asintomáticas o presentarse como un cuadro inespecífico de dolor abdominal, vómitos y diarrea, no siempre asociando ictericia. Es importante vigilar la evolución clínica. Una complicación grave de la hepatitis viral es la hemofagocitosis linfohistiocitósica secundaria a VEB y, en ocasiones, el fallo hepático agudo.
Genética
En la actualidad, gracias a los avances en investigación, se incluyen las pruebas genéticas en escalones avanzados del proceso diagnóstico. El estudio de un gen en la sospecha de enfermedad nos permite evitar técnicas más invasivas, como biopsia de hígado, piel o músculo. También son pruebas muy importantes para el estudio de familiares (hermanos, padres), por ejemplo, en déficit de alfa-1 antitripsina o enfermedad de Wilson.
En casos en los que no es fácil dilucidar el origen de la hepatopatía, se emplean paneles genéticos específicos de enfermedad hepática o que implican alteración en el hígado: transporte de ácidos biliares, fallo hepático agudo recurrente, fibrosis hepática por ciliopatías, errores congénitos del metabolismo o colestasis intrahepática familiar, entre otros.
Técnicas de imagen
• Ecografía: identifica malformaciones de la vía biliar o litiasis y tumores, o detecta signos de cronicidad, como ascitis, alteración en la ecogenicidad hepática, esplenomegalia, presencia de varices o alteración en los flujos arterial o venoso en el estudio Doppler.
• Colangioresonancia: en la patología autoinmune hepática se debe realizar para descartar afectación de vía biliar (colangitis esclerosante aislada o síndrome de solapamiento).
• Elastografía: técnica que informa del grado de fibrosis del hígado y de la proporción de grasa. La velocidad de propagación de la onda elástica es proporcional a la rigidez. Tiene buena correlación con los resultados obtenidos por biopsia hepática(14).
Técnicas invasivas
• Biopsia hepática: la anatomía patológica puede ayudar a diferenciar ciertas patologías: hepatitis autoinmune, donde se detectará hepatitis de interfase e infiltrado de células plasmáticas; enfermedad de Wilson, con cuantificación de cobre en tejido; defecto de alfa-1 antitripsina, con hallazgo de gránulos PAS positivo; o atresia de vía biliar, con colestasis y proliferación ductal. Con técnicas de inmunohistoquímica, se puede detectar la presencia o ausencia de transportadores del canalículo biliar y proteínas de membrana, permitiendo diagnosticar colestasis intrahepáticas familiares (BSEP, MDR3).
• Endoscopia: en niños con hipertensión portal avanzada con marcada trombopenia, se realiza un programa de profilaxis primaria de sangrado con endoscopia y tratamiento en caso de hallazgo de varices (colocación de bandas).
• Colangiografía intraoperatoria: gold standard en el diagnóstico de la atresia de vía biliar, consiste en inyectar contraste en la vía biliar, permitiendo delimitar su anatomía. La colangiografía también puede servir como medida terapéutica en los casos de colestasis obstructiva o lactantes con síndrome de bilis espesa o litiasis.
• Colangiografía transparietohepática: punción ecoguiada del parénquima hepático realizada por radiólogos intervencionistas, para, a través de un radical biliar periférico, inyectar contraste y llegar a la vía biliar principal. Permite el tratamiento de complicaciones, principalmente en pacientes trasplantados: dilatación con balón y/o colocación de stent.
Función del pediatra en Atención Primaria
El pediatra de Atención Primaria será quien, muchas veces, manteniendo un alto grado de sospecha, sea capaz de diagnosticar un paciente con hepatopatía, como es el caso de una ictericia prolongada en un lactante. Al mes de vida, hasta 1/5 de los neonatos estarán ictéricos por hiperbilirrubinemia indirecta en relación con lactancia materna, pero es importante descartar enfermedad hepática. A partir de los 15 días de vida se debe solicitar una analítica, discriminando entre bilirrubina directa e indirecta para su derivación precoz y ver las deposiciones en persona para asegurarse de la ausencia de acolia o hipocolia, ya que la descripción por parte de la familia puede ser errónea (v. Algoritmo 1 al final del artículo).
Igualmente, realizará el seguimiento de pacientes con hipertransaminasemia hasta que la analítica sea normal; en caso de no resolverse, se debe ampliar estudio y, si es necesario, derivar a un especialista en gastroenterología pediátrica o hepatología (v. Algoritmo 2 al final del artículo).
Los signos de alarma que se deben tener presentes son: encefalopatía, coagulopatía que no revierte con vitamina K, otros signos de insuficiencia hepática, como hipoalbuminemia o hipoglucemia, o desarrollo de hipertensión portal (esplenomegalia o trombopenia).
Conflicto de intereses
No hay conflicto de interés en la elaboración del presente manuscrito ni fuente de financiación.
Bibliografía
Los asteriscos muestran el interés del artículo a juicio de las autoras.
1.* Jara P. Enfermedad hepática en el niño. Tile Von Spain. S.L. 2013.
2.* Lledín Barbancho D, Vecino López R. Exploraciones clínicas, bioquímicas y técnicas de imagen en la valoración de la patología digestiva y hepatobiliar. Pediatr Integral. 2015; XIX: 66.e1-e18. Disponible en: https://www.pediatriaintegral.es/publicacion-2015-01/exploraciones-clinicas-bioquimicas-y-tecnicas-de-imagen-en-la-valoracion-de-la-patologia-digestiva-y-hepatobiliar/.
3. Lee Ng V. Laboratory Assessment of Liver Function and Injury in Children. En: Suchy FJ, Sokol RJ, Balistreri WF, eds. Liver Disease in Children. Cambridge University Press; 2007. p. 163-76.
4. Vos MB, Abrams SH, Barlow SE, Caprio S, Daniels SR, Kohli R, et al. NASPGHAN Clinical Practice Guideline for the Diagnosis and Treatment of Nonalcoholic Fatty Liver Disease in Children: Recommendations from the Expert Committee on NAFLD (ECON) and the North American Society of Pediatric Gastroenterology, Hepatology and Nutrition (NASPGHAN). J Pediatr Gastroenterol Nutr. 2017; 64: 319-34.
5. Johansen L, BSPGHAN Liver Steering Group. UK Fatty Liver Disease Guideline. 2020.
6.* Hegarty R, Dhawan A. Fifteen-minute consultation: The child with an incidental finding of elevated aminotransferases. Arch Dis Child Educ Pract Ed. 2018; 103: 228-30.
7.* Squires JE, McKiernan P, Squires RH. Acute Liver Failure: An Update. Clin Liver Dis. 2018; 22: 773-805.
8. Baker A, BSPGHAN. Investigation and treatment of liver disease with acute onset. Local hospital protocol. 2013.
9. Fernández Tomé L, Frauca Remacha E. Colestasis en el lactante. Protoc diagn ter pediatr. 2023; 1: 341-60.
10. Maisels MJ, Clune S, Coleman K, Gendelman B, Kendall A, McManus S, et al. The natural history of jaundice in predominantly breastfed infants. Pediatrics. 2014; 134: e340-5.
11. Fawaz R, Baumann U, Ekong U, Fischler B, Hadzic N, Mack CL, et al. Guideline for the evaluation of cholestatic jaundice in infants: joint recommendations of the North American Society for Pediatric Gastroenterology, Hepatology and Nutrition and the European Society for Pediatric Gastroenterology, Hepatology and Nutrition. JPGN. 2017; 64: 154-68.
12. Heiduk M, Päge I, Kliem C, Abicht K, Klein G. Pediatric reference intervals determined in ambulatory and hospitalized children and juveniles. Clin Chim Acta. 2009; 406: 156-61.
13. Mieli-Vergani G, Vergani D, Baumann U, Czubkowski P, Debray D, Dezsofi A, et al. Diagnosis and Management of Pediatric Autoimmune Liver Disease: ESPGHAN Hepatology Committee Position Statement. J Pediatr Gastroenterol Nutr. 2018; 66: 345-60.
14. Levitte S, Lee LW, Isaacson J, Zucker EJ, Milla C, Barth RA, et al. Clinical use of shear-wave elastography for detecting liver fibrosis in children and adolescents with cystic fibrosis. Pediatr Radiol. 2021; 51: 1369-77.
15. Ros Arnala I, Reyes Andrade J, Mercadal Hally M, Blesa Baviera LC, García Tirado D, Campuzano Martín SH, et al. Actuación diagnóstica ante hipertransaminasemia en pediatría: documento de consenso de Sociedad Española de Gastroenterología, Hepatología y Nutrición Pediátrica (SEGHNP), Asociación Española de Pediatría de Atención Primaria (AEPap) y Sociedad Española de Pediatría de Atención Primaria (SEPEAP). Anales de Pediatría. 2022; 96: 448.e1-e11.
Bibliografía recomendada
– Jara P. Enfermedad hepática en el niño. Tile Von Spain. S.L. 2013.
Manual de fácil lectura que sintetiza la patología hepática pediátrica y las indicaciones y complicaciones del trasplante hepático. Algunos avances terapéuticos, dado el año de publicación, no están incluidos, pero cuenta con grandes autores.
– Lledín Barbancho D, Vecino López R. Exploraciones clínicas, bioquímicas y técnicas de imagen en la valoración de la patología digestiva y hepatobiliar. Pediatr Integral. 2015; XIX: 66.e1-e18. Disponible en: https://www.pediatriaintegral.es/publicacion-2015-01/exploraciones-clinicas-bioquimicas-y-tecnicas-de-imagen-en-la-valoracion-de-la-patologia-digestiva-y-hepatobiliar/.
Completo y clarificador artículo sobre la aproximación diagnóstica a un niño con sospecha de enfermedad hepática.
– Hegarty R, Dhawan A. Fifteen-minute consultation: The child with an incidental finding of elevated aminotransferases. Arch Dis Child Educ Pract Ed. 2018; 103: 228-30.
Breve y conciso artículo del centro de referencia inglés King’s College Hospital sobre el manejo en Atención Primaria de un niño con posible hepatopatía, sintetizando los datos de alarma.
– Squires JE, McKiernan P, Squires RH. Acute Liver Failure: An Update. Clin Liver Dis. 2018; 22: 773-805.
Resumen del mayor registro de fallo hepático agudo en niños recogido, actualizando los protocolos diagnósticos con recomendaciones para optimizar el manejo y derivación precoz al centro de trasplante.
| Caso clínico |
|
Niño de 10 años que acude tras cuadro febril de 5 días de evolución, refiere dolor abdominal leve y sensación de astenia. Destaca en la exploración física: arañas vasculares en manos y esplenomegalia de tres traveses; peso: 35 kg p45; talla: 140 cm p51; IMC: 17,8 kg/m2. Últimamente, ha tenido varias epistaxis y se han citado en otorrino privado, pero todavía no ha tenido la consulta. Analítica en Urgencias: AST: 259 UI/L; ALT: 122 UI/L; GGT: 44 UI/L; bilirrubina total: 1,2 mg/dL; actividad de protrombina: 55 %; plaquetas: 40 x 10 e3/µL. Destaca en la exploración: distensión abdominal, esplenomegalia, circulación colateral y spiders. Antecedentes personales: refieren que hace 2 años hicieron una primera analítica en Urgencias con hallazgo: AST: 313 UI/L; ALT: 602 UI/L; GGT: 71 UI/L; bilirrubina total: 0,6 mg/dL; directa: 0,2 mg/dL; plaquetas: 300 x 10 e3/µL. No extraída coagulación. Al repetir el control a los 10 días en su centro de salud: AST: 199 UI/L; ALT: 229 UI/L; GGT: 49 UI/L; bilirrubina total: 0,7 mg/dL; directa: 0,2 mg/dL; actividad de protrombina: 90 %; plaquetas: 280 x 10 e3/µL y hallazgo de serología de parvovirus IgM positiva, pero no volvieron a hacer nuevos controles. Antecedentes familiares: abuelo materno con artritis reumatoide y abuela paterna con diabetes mellitus tipo 2, resto sin interés.
|


 Diagnosis in liver disease
Diagnosis in liver disease