 |
| Temas de FC |
C. Camarena Grande
Servicio de Hepatología y Trasplante Hepático Pediátrico. Hospital Infantil La Paz, Madrid
| Resumen
La incidencia de hepatitis viral en la infancia ha disminuido en nuestro medio, debido a: la vacunación de la hepatitis B, la mejoría de las condiciones higiénicas que evitan la hepatitis A y el tratamiento de la hepatitis C que evita la transmisión vertical. Las causas más frecuentes de hepatitis no infecciosa son: la hepatitis autoinmune y la tóxica. Su presentación puede ser similar a una hepatitis aguda viral, por lo que es muy importante la sospecha diagnóstica. En el caso de la hepatitis autoinmune, el diagnóstico y tratamiento precoz evitan la evolución a cirrosis y la necesidad de trasplante hepático. En la hepatitis tóxica, su diagnóstico y suspensión del tratamiento causal puede evitar la progresión del daño y, en algunos casos, como en la hepatitis por acetaminofeno, permite instaurar un tratamiento específico. |
| Abstract
The incidence of viral hepatitis in childhood has decreased in our environment due to hepatitis B vaccination, improvement of hygienic conditions that prevent hepatitis A and treatment of hepatitis C that averts vertical transmission. |
Palabras clave: Hepatitis autoinmune; Hepatitis tóxica.
Key words: Autoimmune hepatitis; toxic hepatitis.
Pediatr Integral 2020; XXIV(1): 28 – 37
Hepatitis no infecciosas
Hepatitis autoinmune
Introducción
La hepatitis autoinmune (HAI) es una enfermedad responsable de la destrucción progresiva del parénquima hepático que evoluciona hacia cirrosis e insuficiencia hepática y que, sin tratamiento, conlleva una elevada mortalidad.
Tiene un curso crónico con fluctuaciones y su comienzo está frecuentemente mal definido. Se caracteriza por: elevación de transaminasas en suero, niveles elevados de inmunoglobulina IgG, presencia de autoanticuerpos circulantes no órgano-específicos e inflamación en la histología, con hepatitis de interfase en ausencia de otra etiología conocida. Predomina en el sexo femenino y suelen coexistir otras enfermedades autoinmunes en el paciente o en sus familiares.
La hepatitis autoinmune representa una interacción compleja entre: factores desencadenantes, mimetismo molecular, predisposición genética y alteración en las redes de inmunorregulación.
La hepatitis autoinmune se clasifica de acuerdo a su perfil serológico en: tipo 1, con positividad para anticuerpos antinucleares (ANA), antimúsculo liso (AML) o ambos; y tipo 2, con positividad para anticuerpos antimicrosoma de hígado-riñón (LKM1) y/o anticitosol hepatico tipo 1 (LC-1). La hepatitis autoinmune tipo 1 afecta a adultos y niños, mientras que la tipo 2 es fundamentalmente pediátrica. Un 20% de pacientes pueden no tener al inicio: ANA, AML o anti-LKM1 (hepatitis autoinmune seronegativa).
Epidemiología
La incidencia anual en Europa es de 1,9/100.000 habitantes y su prevalencia de 16,9/100.000 habitantes. Es responsable del 2,6% de los trasplantes hepáticos en Europa y del 5,9% en EE.UU.
La incidencia es mayor en mujeres que en hombres (3,6:1) y se presenta en todas las edades y grupos étnicos. En una serie pediátrica reciente en Canadá, su incidencia fue de 0,23 casos/100.000 niños/año(1).
En la actualidad, su diagnóstico es más frecuente por: una mayor vigilancia, disminución de la incidencia de hepatitis viral y un aumento real de su incidencia.
Fisiopatología
La lesión hepática se inicia y perpetúa por una respuesta inmune frente a autoantígenos hepáticos con fallo en los mecanismos inmunorreguladores.
El daño hepático es causado por linfocitos T CD4, aunque probablemente estén involucradas otras poblaciones celulares, incluyendo a las células Th17. En el infiltrado portal, hay un reclutamiento masivo de células inflamatorias activadas que causan el daño, sobre todo: linfocitos T CD4, pocos CD8, células NK, macrófagos, células B y células plasmáticas. Los linfocitos CD4 reconocen autoantígenos peptídicos. En presencia de señales co-estimulatorias apropiadas, se inicia la respuesta inmune después de que el péptido sea abrazado por una molécula HLA de clase II y presentado por una célula presentadora de antígeno (APC) a un linfocito naïve T-helper CD4+ (Th0). Una vez activadas las células Th0 y, dependiendo del tipo de citoquinas presentes y de la naturaleza del antígeno, se pueden diferenciar en células: Th1, Th2 y Th17. Estas células efectoras inician una cascada de reacciones inmunes determinada por las citoquinas que producen:
• Las células Th1 producen IL-2 e IF gamma, que es considerado el principal causante del daño hepático: al estimular las células CD8, aumentar la expresión de moléculas HLA-1 e inducir la expresión de las de clase HLA-2 en los hepatocitos, también activa los monocitos/macrófagos que, a su vez, liberan IL-1 y TNF-alfa.
• Las células Th2 producen IL-4, 10 y 13, citoquinas que inducen la maduración de las células B en células plasmáticas, con la consecuente producción de anticuerpos.
• Células Th17, que en presencia de TGF-beta e IL-6, producen: IL-17, IL-22, TNF-alfa y el ligando de quimoquina CCL-20. Las células Th17 se encuentran aumentadas en sangre e hígado de los pacientes con HAI. La destrucción de células hepáticas se perpetúa por alteración funcional y el número de las células T reguladoras (CD24, CD25)(2).
Entre los factores ambientales desencadenantes se han propuesto: fármacos (halotano, hidralacina, minociclina) y virus (VHA, sarampión, EBV, HSV).
La mayor comprensión de los mecanismos involucrados en la patogénesis contribuirá al desarrollo de nuevos tratamientos como: la transferencia de linfocitos T reguladores autólogos, expandidos, específicos de antígeno para restaurar la tolerancia a antígenos hepáticos.
Genética
La hepatitis autoinmune tiene asociación con el locus (HLA)-DRB1 del antígeno leucocitario humano, el HLA-DRB*03 y el HLA-DRB*04 confieren susceptibilidad en las poblaciones europeas y norteamericanas.
La susceptibilidad para la HAI-2 se asocia con HLA-DR*03 y HLA-DR*07 en Reino Unido y Brasil, así como a DQB1*02. Otras asociaciones no HLA son: CTLA-4 (antígeno-4 asociado al linfocito T citotóxico), el gen promotor de factor de necrosis tumoral TNF-alfa, y el FAS, un importante inductor de la vía extrínseca de la apoptosis.
Criterios diagnósticos de hepatitis autoinmune
Requiere hallazgos característicos: elevación de transaminasas y de IgG, autoanticuerpos positivos y hepatitis de interfase, junto con la exclusión de otras enfermedades que recuerden a la hepatitis autoinmune(3): enfermedad de Wilson, déficit de alfa-1-antitripsina, infección viral (virus de la hepatitis A, B, C) y fármacos (minociclina, nitrofurantoína, isoniazida, propiltiouracilo, alfa-metil-dopa) (Tabla I).
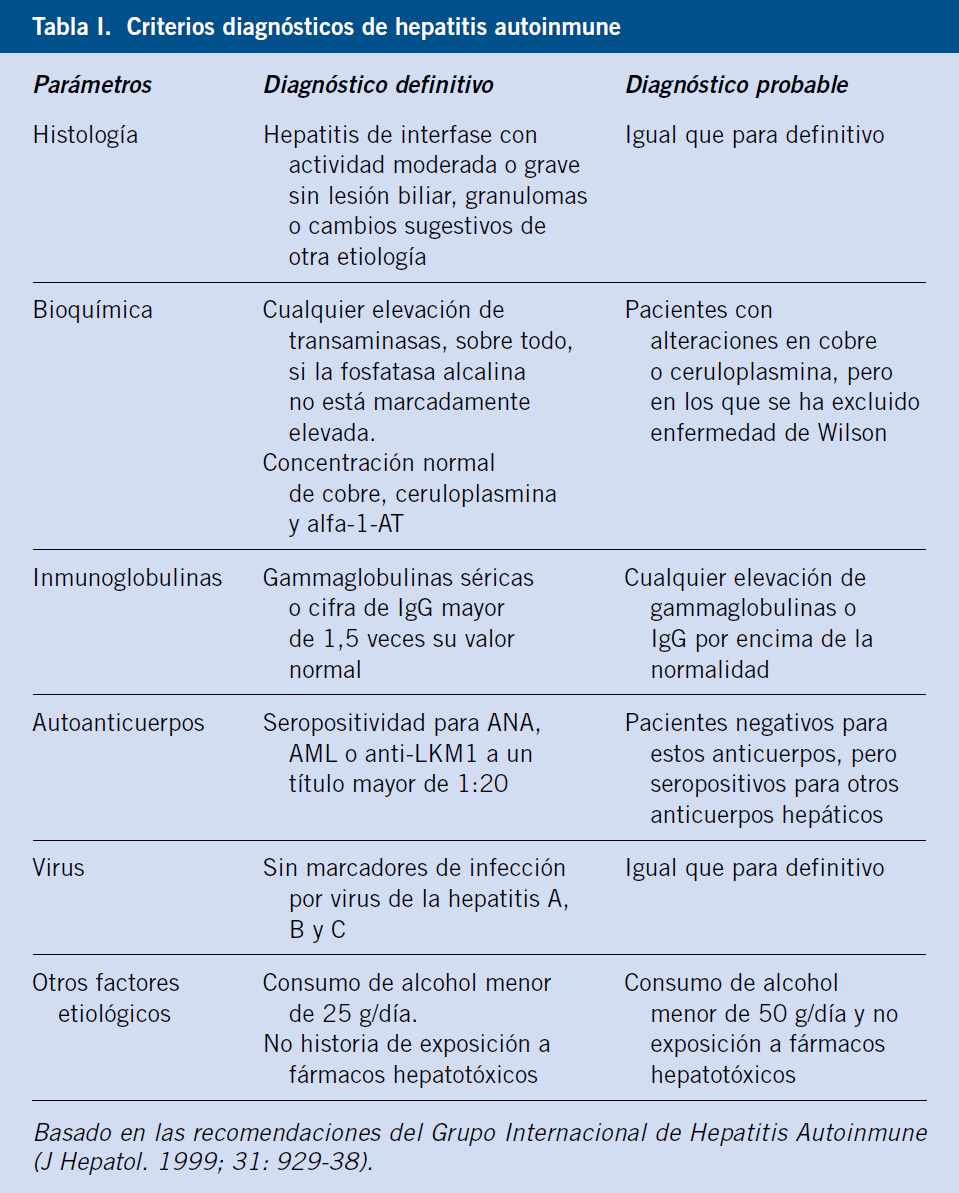
En los años 90, hubo dos reuniones del grupo internacional de expertos en hepatitis autoinmune que desarrollaron un sistema de “score” para su diagnóstico(4). La diferencia entre un diagnóstico definitivo y probable se relaciona con el grado de elevación de IgG y anticuerpos. La presencia de datos de laboratorio e histológicos de colestasis conllevan un score negativo. En ausencia de anticuerpos convencionales, la presencia de autoanticuerpos: frente al receptor de asialoglicoproteína (anti-ASGPR); contra el antígeno citosólico hepático tipo 1 (anti-LC-1); contra el antígeno soluble hígado/páncreas (anti-SLA/LP); y anticitoplasma de neutrófilo perinuclear atípico, sostienen el diagnóstico. La respuesta a tratamiento con esteroides fortalece el diagnóstico de HAI. El diagnóstico de hepatitis autoinmune es definitivo con una puntuación mayor de 15 antes de tratamiento o mayor de 17 después de este. La sensibilidad del score es del 97-100% (Tabla II).

Los criterios diagnósticos en niños son diferentes que en adultos, los niveles de autoanticuerpos tienden a ser menores y su presencia a cualquier título en combinación con otro elemento requerido, es suficiente para el diagnóstico.
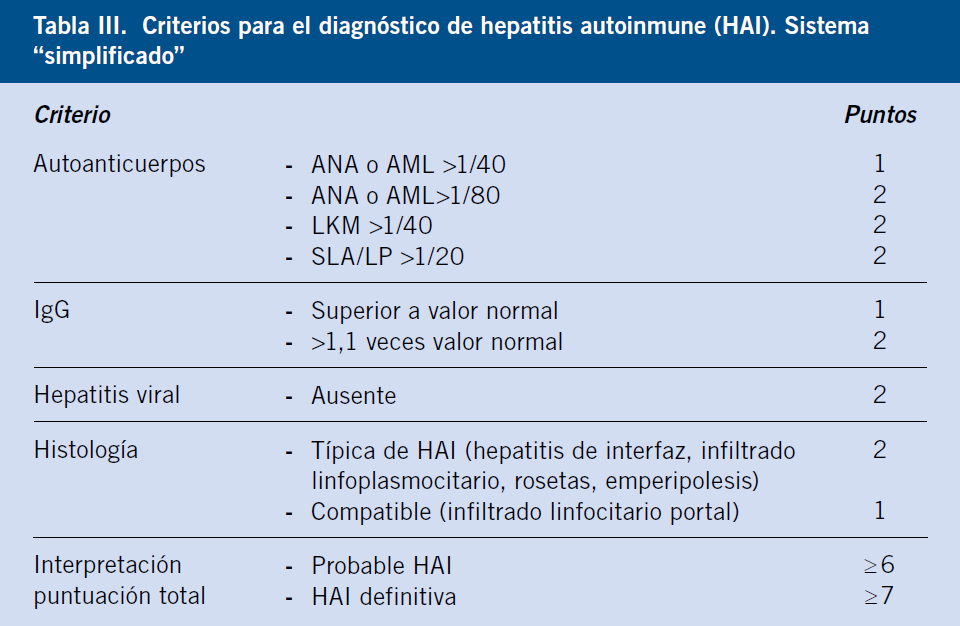
Posteriormente, se ha propuesto un score simplificado para uso clínico, que solo utiliza 4 criterios: autoanticuerpos, IgG, histología y exclusión de hepatitis viral. Ofrece datos clave para su sospecha, de forma que una mayoría de médicos los conozcan y orienten al paciente a un tratamiento precoz. No ha probado su utilidad en niños, debido a su baja sensibilidad, de forma que solo diagnosticaría al 20% de casos de HAI fulminante(5) (Tabla III).
Clínica
Se debe sospechar en un niño con síntomas agudos o crónicos de hepatitis en ausencia de otras etiologías, sobre todo, si en él o en sus familiares coexisten enfermedades autoinmunes.
Más de la mitad de los niños presentan al inicio una clínica similar a una hepatitis aguda ictérica prolongada, un 10% (sobre todo, pacientes LKM1 y menores de 2 años) debutan como fallo hepático, el resto inician una clínica insidiosa de enfermedad hepática y un pequeño porcentaje son diagnosticados de forma casual(3,6,7). Excluidos los niños con fallo hepático, la clínica es similar en la tipo 1 y en la tipo 2. En la exploración física de los pacientes, es frecuente apreciar estigmas de hepatopatía crónica a pesar de una presentación aguda.
Histología
La diana de la lesión es el hepatocito. Por tanto, existirá hepatitis con actividad moderada o grave con necrosis periportal, conocida como hepatitis de interfase, con o sin hepatitis lobular o necrosis en puentes centro-portal, siendo el infiltrado portal predominantemente linfoplasmocitario.
No deben existir lesiones biliares, granulomas, depósitos de cobre u otros cambios que sugieran otra etiología. Los hallazgos de ductopenia o colangitis destructiva añadida pueden apoyar el diagnóstico de colangitis esclerosante autoinmune descrito en niños. En niños con HAI-1, se recomienda realizar colangiorresonancia magnético-nuclear para descartar colangitis autoinmune. La presencia de esteatosis o sobrecarga de hierro sugieren diagnósticos alternativos como: hepatitis C, enfermedad de Wilson o toxicidad por drogas. Existe fibrosis en prácticamente todos los pacientes. La histología inicial es de cirrosis, en la mitad de los niños (Fig. 1).
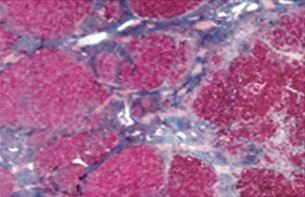
Figura 1. Cirrosis en hepatitis autoinmune. Fibrosis confluente que delimita nódulos de regeneración.
Los niños con fallo hepático presentan colapso multi o panacinar(2,3,7) (Fig. 2).
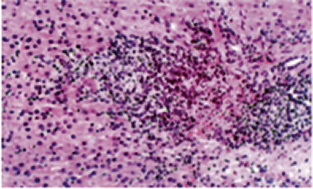
Figura 2. Hepatitis periportal, intenso infiltrado inflamatorio en espacio porta.
Bioquímica
Puede existir cualquier elevación de transaminasas, que suele ser importante, sin elevación marcada de la fosfatasa alcalina ni GGT. La concentración de gammaglobulinas séricas o IgG total debe ser mayor de 1,5 veces su límite superior. Hasta un 20% de niños no muestran elevación de gammaglobulinas.
Los niveles de transaminasas y gammaglobulina no predicen el patrón histológico ni la presencia de cirrosis. Las concentraciones de cobre, ceruloplasmina y alfa-1-antitripsina deben ser normales(3,4).
Casi la mitad de los niños presenta datos de insuficiencia hepática (INR alargado y disminución de albúmina) al inicio de la enfermedad. Se suelen asociar otras alteraciones inmunológicas, como déficit de IgA (hasta en el 40% de niños con tipo 2), de C3 y de C4.
Autoanticuerpos
Seropositividad de ANA, AML a título mayor de 1:20 y LKM, y anti LC-1 mayor de 1:10 en niños mediante inmunofluorescencia indirecta en sustrato fresco de roedores, que incluye: riñón, hígado y estómago.
Otros autoanticuerpos realizados menos comúnmente, pero de importancia diagnóstica son: antiantígenosoluble hepático (anti SLA) que identifica a pacientes con curso más severo y p-ANCA (anticuerpos contra el citoplasma de los neutrófilos con patrón perinuclear) asociado con frecuencia a enfermedad inflamatoria intestinal y colangitis esclerosante autoinmune(3,4). Los autoanticuerpos no son patogénicos ni específicos de la enfermedad y su expresión puede variar en el curso de la hepatitis autoinmune. Un título bajo no excluye el diagnóstico ni uno alto lo confirma en ausencia de otros datos. En niños se ha demostrado, en la evolución de la hepatitis autoinmune, correlación entre el grado de actividad de la enfermedad y el nivel de IgG y autoanticuerpos. Los pacientes seronegativos al inicio pueden ser clasificados como hepatitis criptogenética hasta que aparezcan marcadores convencionales en la evolución u otros autoanticuerpos, que no están disponibles convencionalmente(3,8).
Subclasificaciones
Se han propuesto dos tipos según los marcadores inmunológicos. No tienen distinta etiología ni respuesta a tratamiento en niños mayores.
Hepatitis autoinmune tipo 1
Se asocia con ANA/anti-ML. Constituye el 60% de casos en niños. Afecta a todas las edades y se asocia con HLA DR3 (DRB1*0301) y DR4 (DRB1*0401) en caucásicos de Europa y norteamericanos. Los DR3 y DR4 influencian la expresión de la enfermedad, su comportamiento y susceptibilidad.
Los pacientes con DR3 son más jóvenes, presentan mayor falta de respuesta al tratamiento y a las recaídas, y necesitan más frecuentemente trasplante que los DR4, que son mayores, responden mejor a tratamiento y suelen tener otras enfermedades autoinmunes asociadas. Los pANCA, que son comunes en la HAI tipo 1, no se detectan en la tipo 2.
Hepatitis autoinmune tipo 2
Se caracteriza por la presencia de anti-LKM1, es más frecuente en Europa, donde causa el 40% de casos de hepatitis autoinmune, y Sudamérica, que en América del Norte, y se asocia a DRB1*0701.
Hepatitis autoinmune asociada a enfermedad celiaca
La prevalencia de enfermedad celiaca en niños con HAI es del 19% y muchas veces es seronegativa en ellos.
Indicaciones de tratamiento
Los ensayos controlados han demostrado mejoría clínica, histológica y aumento de supervivencia con tratamiento con corticoides.
Los pacientes con cirrosis responden igual de bien que los no cirróticos y la esperanza de vida a los 20 años en pacientes tratados es mayor del 80%. El tratamiento se debe iniciar en cuanto se realice el diagnóstico(3).
Definición de respuesta al tratamiento
Respuesta completa
Es la mejoría marcada de síntomas y vuelta a la normalidad completa de todos los parámetros de función hepática y gammaglobulinas en el primer año y mantenida, al menos, durante 6 meses de terapia de mantenimiento o, si se ha realizado una biopsia en este período, con actividad mínima.
Recaída
Es el restablecimiento de la actividad de la enfermedad después de la inducción de remisión, bien al suspender el tratamiento o intentar reducirlo. Los pacientes con recaída muestran mayor progresión a cirrosis.
Fallo del tratamiento
Es el empeoramiento clínico y de laboratorio a pesar del tratamiento. Se produce en menos del 9% de los niños. En un 13% de los pacientes ocurre una respuesta incompleta, caracterizada por mejoría, clínica, de laboratorio e histológica, pero sin lograr la remisión.
Tratamiento convencional
La pauta inicial de tratamiento consiste en prednisona (o prednisolona) a dosis de 2 mg/kg/día, con dosis tope de 60 mg/día, durante 15 días. Si se obtiene una buena respuesta, se asocia azatioprina a dosis 1-2 mg/kg y se desciende la dosis de prednisona hasta llegar a 0,1-0,2 mg/kg/día en 6-8 semanas. Posteriormente, si la función hepática es normal, se pasa a administración de prednisona en días alternos, manteniendo fija la azatioprina. La monoterapia con prednisona está indicada en niños con citopenias graves asociadas o no a hiperesplenismo. Se deben realizar controles rutinarios de función hepática, hemograma y amilasa cada 4 semanas hasta la remisión, y luego trimestralmente(3).
Los niños tienen buena respuesta al tratamiento, con normalización de la función hepática en 6-9 meses en el 80%. Son factores pronóstico-relacionados con evolución hacia el fallecimiento o necesidad de trasplante: menor edad al diagnóstico, presencia de LKM1, tiempo de protrombina prolongado, mayor cifra de bilirrubina y mayor índice de actividad histológica. De forma que los niños con hepatopatía grave inicial consiguen la remisión en un 75% y casi el 100% de aquellos con hepatopatía no grave.
El objetivo en los niños, es mantener transaminasas y gammaglobulinas normales o con mínima alteración con la menor dosis posible de medicación. A largo plazo, se suelen conseguir dosis bajas y el tratamiento debe ser individualizado. Tras cinco años de remisión, en pacientes con tratamiento combinado, se puede suspender la prednisona y mantener monoterapia con azatioprina.
El punto final de tratamiento sería cuando se ha conseguido una función hepática normal mantenida durante 2 años y la biopsia hepática no muestra inflamación. Hay que tener en cuenta que en el 55% de los pacientes con transaminasas normales y gammaglobulina durante el tratamiento, se encuentra en la biopsia hepatitis de interfase, y en ellos no se debe retirar el tratamiento. En nuestra experiencia, no se logró la retirada de tratamiento en ningún paciente. En la serie del King’s College, se pudo retirar el tratamiento en un 19% de niños con hepatitis autoinmune tipo 1 y en ninguno con tipo 2(6). Los niños con enfermedad celiaca asociada y que hacen dieta sin gluten pueden tener un riesgo menor de recaída tras la retirada del tratamiento.
En el seguimiento, el 40% de los niños presentan uno o más episodios de recaída (más frecuente en adolescentes por incumplimiento terapéutico) que puede condicionar en un cuarto de niños: presentación grave, evolución hacia insuficiencia hepática y aparición de complicaciones de cirrosis. El tratamiento de la recaída será volver a la pauta original de inmunosupresión y posterior mantenimiento, de forma indefinida.
Los efectos secundarios de los corticoides se observan con mayor frecuencia en niños cirróticos al inicio (que constituyen el 50% de pacientes), posiblemente por mayores niveles en suero de prednisona no ligada por hipoalbuminemia.
La azatioprina puede producir en menos del 10% de los niños, la supresión de médula ósea (leucopenia y trombocitopenia) y también con menor frecuencia: vómitos, hepatitis colestásica, enfermedad venoclusiva, pancreatitis y rash. La eliminación de la 6-mercaptopurina (metabolito activo derivado de la azatioprina) la realiza la tiopurina metiltransferasa; los genes que la codifican son muy polimórficos y la actividad del enzima es inducible por azatioprina. Un 0,3% de la población tiene actividad baja de este enzima y es intolerante a la azatioprina; un 11% de la población son heterocigotos para el déficit enzimático y tienen actividad intermedia. En niñas adolescentes y con riesgo teórico de embarazo, hay que avisar del posible riesgo (solo probado en ratones) de teratogenicidad de la azatioprina: anomalías del esqueleto, paladar hendido, disminución del tamaño del timo, hidrops fetalis, supresión hematopoyética y mayor riesgo de prematuridad y bajo peso neonatal.
Tratamientos alternativos
• Budesonida: es un corticoide de segunda generación, cuyos metabolitos carecen de actividad glucocorticoide. Se ha usado en Europa, en estudios piloto con buenos resultados.
• La ciclosporina, que inhibe la calcineurina e impide la transcripción de la interleucina 2, reduce la expresión de citocinas y disminuye la proliferación de linfocitos T. Se ha demostrado su eficacia y falta de efectos adversos importantes en tratamientos cortos. Se debe monitorizar la función renal y tensión arterial. Solo debe usarse en centros con experiencia en el empleo de este fármaco, ya que en tratamientos prolongados, puede inducir la aparición de síndrome linfoproliferativo relacionado con infección por virus de Epstein-Barr y producir disfunción renal permanente. Se ha usado con éxito en las siguientes situaciones:
- Inducción de remisión en monoterapia con ciclosporina A durante 6 meses, para evitar los efectos adversos de los corticoides a altas dosis en la fase inicial de tratamiento. Posteriormente, se añaden prednisona y azatioprina a dosis bajas, y se suspende la ciclosporina a lo largo del séptimo mes. Este estudio se realizó en 84 niños, se obtuvo la remisión en el 94,1% y los efectos secundarios de la ciclosporina fueron bien tolerados y desaparecieron cuando se suspendió esta. Un paciente precisó trasplante por falta de respuesta(9).
- Tratamiento indefinido en monoterapia con ciclosporina, en niños con riesgo elevado de complicaciones por esteroides (diabetes, cataratas, obesidad) o que han desarrollado esas complicaciones, debido al uso previo de prednisona. El estudio se realizó en 20 niños y todos normalizaron las transaminasas al sexto mes, con mínimos efectos secundarios(3).
- Tratamiento de niños sin remisión con tratamiento convencional, en los que se emplea asociada a prednisona y azatioprina(3).
- Tratamiento de niños con presentación como fallo hepático agudo, en los que se utiliza a dosis de 1 mg/kg/día cada 12 horas por vía intravenosa inicialmente y posterior paso a vía oral, ajustada según niveles, más azatioprina a 1,5 mg/kg/día, más prednisona a 2 mg/kg/día(3).
• El mofetil micofenolato antagoniza la síntesis de purinas y depleciona las reservas de nucleótidos de guanina necesarios para la síntesis de ADN y la expansión de clones de células T. Parece más selectivo para linfocitos que la azatioprina, pero no ha sido probado en estudios amplios en niños. En nuestra experiencia, se ha mostrado útil para conseguir la remisión de la enfermedad en niños con refractariedad al tratamiento convencional y, en casos de intolerancia a azatioprina, con ahorro de esteroides.
• También, se ha probado tratamiento con biológicos en pacientes con mala respuesta: anticuerpo monoclonal frente a células B, Rituximab(3).
Tratamiento de la hepatitis autoinmune refractaria
En un 15% de los niños se produce fallo del tratamiento, bien por falta de remisión inicial o bien por desarrollo de complicaciones de la cirrosis en la evolución (ascitis, sangrado, etc.). Estos niños deben ser valorados para la realización de un trasplante hepático. La HAI es responsable del 2-3% de los trasplantes pediátricos en Europa y Estados Unidos.
En niños con un inicio grave de la enfermedad, se puede determinar en las dos primeras semanas, la probabilidad de una respuesta significativa al tratamiento con corticoides. Un caso especial son los niños que debutan como fallo hepático; un 50-75% son refractarios al tratamiento y precisarán trasplante. Los factores que predicen respuesta a corticoides son: MELD (acrónimo: Model for End-stage Liver Disease) al ingreso <28, ausencia de necrosis masiva, estabilización o mejoría de la bilirrubina e INR en los primeros cuatro días de tratamiento.
La recaída de la enfermedad postrasplante es más frecuente que en adultos y más grave, condicionando frecuentemente la pérdida de injertos. Esta se produce en un 20-30% de niños y la recaída histológica puede preceder a la bioquímica. Estos niños presentan, así mismo, mayor gravedad de los episodios de rechazo y de complicaciones vasculares tardías. La supervivencia a 5 años del trasplante es del 86%(3).
Hepatitis tóxica
Introducción
Una de las funciones primarias del hígado es la biotransformación de medicinas, suplementos y productos de herbolario en compuestos, que puedan ser metabolizados y excretados de forma segura por el organismo. Cuando esto no sucede así, se produce daño hepático por drogas que puede ser: predecible y dosis dependiente, como es el caso del acetaminofeno, o indiosincrásico, que requiere un alto índice de sospecha para realizar el diagnóstico y retirar el agente causal.
Epidemiología
La incidencia de daño hepático por drogas en adultos es de 14 casos/100.000 habitantes/año.
En niños es desconocida, se piensa que es menor, porque los niños toman menos medicaciones, es menos probable que abusen del alcohol y que fumen (los factores alteran el metabolismo), se les prescriben menos medicaciones asociadas a la toxicidad por drogas y tienen un metabolismo diferente. El 19% de los casos de fallo hepático agudo en niños son producidos por toxicidad severa por drogas, siendo el paracetamol el más frecuente con un 14% de casos(10).
Etiología
Se han asociado más de 1.000 medicaciones, hierbas y suplementos con hepatotoxicidad. En EE.UU. e Inglaterra, la causa más frecuente es el acetaminofeno.
En nuestro medio existe un pico de hepatitis por acetoaminofeno en niñas adolescentes con fines autolíticos; en ellas, es muy importante un alto índice de sospecha ante clínica sugestiva, dada la tendencia a ocultar la ingesta y la necesidad de un tratamiento precoz.
En el estudio pediátrico prospectivo DILIN(11), los antimicrobianos causaron el 50% de los casos y los agentes con acción sobre el sistema nervioso central el 40%. La minociclina para el acné fue la medicación más frecuente, 13% de casos de hepatotoxicidad. De los fármacos con acción sobre el SNC: 20% fueron antiepilépticos (lamotrigina, valproato(12) y fenobarbital), 13% fármacos empleados para la hiperactividad-déficit de atención (atomoxetina, metilfenidato) y 7% antidepresivos (fluoxetina, amitriptilina y perfenacina) (Tabla IV).

En la base de datos de la OMS(13): los antibióticos (oxacilina y minociclina), los agentes que actúan sobre el SNC (pemolina y ácido valproico), antimetabolitos (metrotexate, mercaptopurina) y los antiretrovirales, se asociaron con una mayor frecuencia de hepatotoxicidad en niños que en adultos. Otros agentes recientemente implicados con fallo hepático agudo han sido: metilfenidato, extracto concentrado de té verde y eculizumab (utilizado para el tratamiento de síndrome hemolítico urémico atípico)(14).
En Asia, la hepatotoxicidad por “hierbas” empleadas en medicina alternativa es más frecuente y sería, cercana a 12 casos/100.000 habitantes año(16), aunque es difícil precisar que producto la causa, debido a que muchas veces se emplean muchas hierbas o constituyentes.
Fisiopatología
El hígado metaboliza y excreta las medicaciones en tres pasos:
• Fase 1 activación: el citocromo P450 inserta un residuo de oxígeno en la medicación, haciéndola más hidrosoluble y a la vez más tóxica.
• Fase 2 detoxificación: mediante conjugación del metabolito, aumenta la hidrosolubilidad y se neutraliza la toxicidad.
• Fase 3: excreción del producto hidrosoluble, se transporta al canalículo y se segrega a la bilis.
La mayoría de las veces, la hepatotoxicidad se produce por acumulación de metabolitos de fase 1, siguiendo un patrón de toxicidad intrínseca o idiosincrásica.
Las drogas con hepatoxicidad intrínseca causan daño hepático previsible dosis dependiente. El paradigma es el acetoaminofeno que, en grandes dosis, se convierte, por medio de las mono-oxigenasas del CYP 450, en NAPQI (N-acetil-p benzoquinona imina); esta es neutralizada por el glutatión, si se sobrepasa la reserva de glutatión, la NAPQI daña los hepatocitos. La toxicidad del acetoaminofeno puede ser revertida por la N-acetilcisteína que restaura la producción de glutatión.
La mayoría de otras drogas causan hepatotoxicidad de forma idiosincrásica, con un periodo de latencia variable. Suelen deberse a variaciones individuales de fases 1 y 2 por: variantes genéticas y epigenéticas, edad, sexo, hormonas, estado nutricional, polimedicaciones, alcohol y tabaco. Así, existen polimorfismos de CYP 450 que condicionan el metabolismo de los sustratos y niveles más bajos en el neonato. Más del 50% de las drogas que producen hepatotoxicidad, es por causar disfunción mitocondrial. Los niños con mutaciones de enzimas mitocondriales tienen susceptibilidad aumentada a este tipo de toxicidad. Los polimorfismos de gen POLG (gen gamma de la DNA polimerasa mitocondrial) se han asociado a fallo hepático por valproato(12).
La toxicidad idiosincrásica puede ser causada también por un mecanismo inmunoalérgico que semeja a un daño autoinmune, es el caso de: minociclina, metildopa, diclofenaco, nitrofurantoína, azitromicina, amoxicilina e isoniazida(11). En estos casos, el metabolito de fase 1 se ligaría a proteínas del hepatocito, creando neoantígenos que, en algunos individuos, producirían una respuesta inmune.
Clínica
La latencia, desde que se tomó la medicación y los síntomas, es difícil de definir, la toxicidad puede ocurrir semanas después de suspenderla. Los pacientes pueden tener un curso subclínico con solo alteraciones de laboratorio. Cuando el daño es fundamentalmente de los hepatocitos, la clínica es muy similar a una hepatitis aguda viral con: náuseas, vómitos, anorexia y elevación de transaminasas. A menudo, la afectación es mixta de hepatocitos y colangiocitos, con clínica de hepatitis y síntomas colestáticos: ictericia y prurito. La enfermedad puede progresar a fallo hepático y cirrosis.
Algunas drogas ocasionan un importante daño endotelial y producen enfermedad venoclusiva con: ictericia, hepatomegalia y ascitis. Cuando los metabolitos inducen una reacción inmunoalérgica puede haber síntomas acompañantes de: fiebre, rash, artralgias y edema facial.
Diagnóstico
El diagnóstico de hepatotoxicidad es a menudo de exclusión, basado en la historia clínica, presentación, laboratorio y evolución.
Hay que descartar: hepatitis viral, esteatohepatitis no alcohólica, hepatitis autoinmune, enfermedad de Wilson, causa metabólica y enfermedad mitocondrial; en los casos que predominan los síntomas colestáticos: colangitis esclerosante primaria y enfermedad pancreáticobiliar.
Las transaminasas suelen estar muy elevadas, con aumento de fosfatasa alcalina, GGT y bilirrubina, si se asocia colestasis.
La biopsia hepática no siempre se realiza y está, sobre todo, orientada a descartar otras causas de enfermedad hepática, ya que no hay hallazgos que sean patognomónicos de hepatotoxicidad. Es común observar necrosis hepatocelular, muchas veces acompañada de esteatosis con infiltrado de eosinófilos, a veces, prominente. En la toxicidad crónica, la histología puede ser semejante a la hepatitis autoinmune.
Tratamiento
Lo más importante del tratamiento es la retirada del agente causal y el tratamiento de soporte. Solo es eficaz el tratamiento con N-acetil-cisteína en el caso del acetaminofeno y la L-carnitina para el valproato.
Se ha probado el tratamiento con corticoides en casos de colestasis progresiva y en curso clínico semejante a hepatitis autoinmune, pero su eficacia no ha sido probada.
El pronóstico es variable en la toxicidad por drogas. En algunos casos, se produce descompensación aguda, siendo responsables en EE.UU. del 20% de los fallos hepáticos agudos en niños (sobre todo, causados por acetoaminofeno)(15). Si se producen datos de fallo hepático: coagulopatía, encefalopatía o hipoglucemia, los niños deben ser referidos rápidamente a un centro donde se realice trasplante hepático. En otros casos, la evolución es a enfermedad hepática crónica, a pesar de la retirada de la droga; esto se produce en el 20% de una serie de niños chinos(16) frente al 7% en el estudio DILIN(13). En el resto de casos, se produce recuperación en un periodo de días a meses. Todos los pacientes con toxicidad por drogas deben ser seguidos hasta que los valores de laboratorio se normalicen y hayan desaparecido los síntomas clínicos.
Bibliografía
Los asteriscos muestran el interés del artículo a juicio del autor.
1. Jiménez-Rivera C, Ling SC, Ahmed N, Yap J, Aglipay M, Barrowman N, et al. Incidence and characteristics of autoinmune hepatitis. Pediatrics. 2015; 136: 1237-48.
2. Liberal R, Vergani D, Mieli-Vergani G. Update on Autoimmune Hepatitis. J Clin Transl Hepatol. 2015; 3: 42-52.
3.*** Mieli-Vergani G, Vergani D, Baumann U, Czubkowski P, Debray D, Dezsofi A, et al. Diagnosis and Management of Pediatric Autoinmune Liver Disease: ESPGHAN Hepatology Committee Pasition Stement. J Pediatr Gastroenterol Nutr. 2018; 66: 345-60.
4.** Álvarez F, Berg PA, Bianchi FB, Burroughs AK, Cancado EL, Chapman RW, et al. International Autoimmune Hepatitis Group Report: review of criteria for diagnosis of autoinmune hepatitis. J Hepatol. 1999; 31: 929-38.
5. Hennes EM, Zeniya M, Czaja AJ, Parés A, Dalekos GN, Krawitt EL, et al. Simplified criteria for the diagnosis of autoinmune hepatitis. Hepatology. 2008; 48: 169-76.
6.** Gregorio GV, Portmann B, Reid F, Donaldson PT, Doherty DG, McCartney M, et al. Autoimmune hepatitis in childhood: a 20-year experience. Hepatology. 1997; 25: 541-7.
7.*** Maggiore G, Nastasio S, Sciveres M. Juvenile autoinmune hepatitis: Spectrum of the disease. World J Hepatol. 2014; 6: 464-76.
8. Gregorio GV, McFarlaneB, Bracken P, Vergani D, Mieli-Vergani G. Organ and non-organ specific autoantibodies titres and IgG levels as markers of disease activity: a longitudinal study in childhood autoimmune liver disease. Autoimmunity. 2002; 35: 515-9.
9. Cuarterolo M, Ciocca M, Cañero Velasco C, Ramonet M, González T, López S, et al. Follow-up of Children With Autoimmune Hepatitis treated With Cyclosporine. J Pediatr Gastroenterol Nutr. 2006; 43: 635-9.
10. Murray KF, Hadzic N, Wirth S. Drug related hepatotoxicity and acute liver failure. J Pediatr Gastroenterol Nutr. 2008; 47: 395-405.
11.** Molleston JP, Fontana RJ, López MJ. Charasteristics of idiosyncratic drug-induced liver injury in children: results from the DILIN prospective study. J Pediatr Gastroenterol Nutr. 2011; 53: 182-89.
12. Mc Farland R, Hudson G, Taylor RW. Reversible valproato hepatotoxicity due to mutations in mitocondrial DNA polymerase gamma (POLG1). Arch Dis Child. 2008; 93: 151-3.
13. Hunt CM, Yuen NA, Stirnadel-FarrantHA, Suzuki A. Age-related differences in reporting of drug-associated liver injury: data-mining of WHO Safety Report Database. Regul Toxicol Pharmacol RTP. 2014; 70: 519-26.
14.*** Amin MD, Harpavat S, Leung DH. Drug-induced liver injury in children. Curr Opin Pediatr. 2015; 27: 625-33.
15.** Squires RH, Shneider BL, Bucuvalas J. Acute liver failure in children: the first 348 patients in the pediatric acute failure study Group. J Pediatr. 2006; 148: 652-8.
16. Zhu Y, Li YG, Wang JB. Causes, features, and outcomes of drug-induced liver injury in 69 children from China. Gut Liver. 2015; 9: 525-33.
Bibliografía recomendada
– Mieli-Vergani G, Vergani D, Baumann U, Czubkowski P, Debray D, Dezsofi A, et al. Diagnosis and Management of Pediatric Autoinmune Liver Disease: ESPGHAN Hepatology Committee Pasition Stement. J Pediatr Gastroenterol Nutr. 2018; 66: 345-60.
Artículo de posicionamiento realizado por un grupo de expertos y los miembros del Comité de Hepatología de la Sociedad Europea de Gastroenterología, Hepatología y Nutrición Pediátrica (ESPGHAN), centrado en el diagnóstico, tratamiento y seguimiento de la enfermedad hepática autoinmune en el niño. Se revisan los artículos publicados en los últimos 30 años.
- Gregorio GV, Portmann B, Reid F, Donaldson PT, Doherty DG, McCartney M, et al. Autoimmune hepatitis in childhood: a 20-year experience. Hepatology. 1997; 25: 541-7.
El valor de este artículo es presentar la experiencia del King’s College Hospital en una amplia serie de niños con hepatitis autoinmune: diagnóstico, respuesta a tratamiento y seguimiento prolongado.
- Amin MD, Harpavat S, Leung DH. Drug-induced liver injury in children. Curr Opin Pediatr. 2015; 27: 625-33.
Revisión de la etiología, epidemiología, fisiopatología y tratamiento de la hepatitis tóxica en niños.
| Caso clínico |
|
Niña de 2 años que acude a urgencias por cuadro de una semana de evolución de: decaimiento, febrícula, anorexia y vómitos ocasionales. Deposiciones diarreicas los tres primeros días, que han pasado a ser pálidas con orina oscura. Antecedentes personales Sin interés, salvo viaje a Marruecos hace dos meses. Antecedentes familiares: madre hipotiroidea tras tiroiditis autoinmune. Exploración física Peso: P25; talla: P50. Algo decaída. Subictericia escleral. ACP: normal. Abdomen ligeramente doloroso a la palpación en hipocondrio derecho, hepatomegalia de 3 traveses de consistencia firme, esplenomegalia de 2 traveses. ORL normal. Neurológico normal. Dos spiders en mano derecha. Pruebas complementarias Hemograma: leucocitos: 12,56 x 10e3/µ (31N, 59L). Hemoglobina: 10* g/dL. Plaquetas 498 x 10e3/µL (180-490). Coagulación: APP: 62%; fibrinógeno: 111 mg/dl; cefalina: normal. Control tras administración de 10 mg de vitamina K IV: APP 58%. Bioquímica: glucosa: 72 mg/dl; AST: 2.843 UI/L; ALT: 1.369 UI/L; GGT: 157 UI/L; colinesterasa: 4.056 UI/L (4.900-11.900); bilirrubina total: 10,3 mg/dl; bilirrubina directa: 7,69 mg/dl; proteínas totales: 7,8g/dl; albúmina: 3,6 g/dl. Serología: VHA IgM (-), VHB: HBsAg (-), anticore (-), anti HBs (+), CMV IgG (+), IgM (-), EBV IgG (-). Inmunología: IgG: 1.990 mg/dL; IgA: 98 mg/dl; IgM: 213 mg/dl; C3: 103mg/dl; C4: 20 mg/dl. ANA negativo: Ac anti músculo liso negativo; ac anti LKM positivo 1/320; anticuerpos antitransglutaminasa IgA negativo. Ecografía abdominal: hepatomegalia, hígado ligeramente heterogéneo, flujo portal 20 cm/s (normal), índice de resistencia de arteria hepática 0,8 (aumentado).
|

