 |
| Regreso a las bases |
D. Infante Pina, P. Ribes Cajas.
Unidad de Gastroenterología y Nutrición Pediátrica. Hospital Universitario General de Catalunya. Grupo Quirón Salud. Barcelona
| Resumen
Se revisa la embriología de la formación del sistema hepatobiliar y se describen los aspectos: clínicos, diagnósticos y terapéuticos de las patologías más frecuentes de presentación neonatal, como: atresia de vías biliares, quiste de colédoco y paucidad de vías biliares intrahepáticas. La presentación típica es de colestasis neonatal con la tríada clásica de: ictericia, acolia y hepatomegalia. Es de suma importancia, el conocimiento de estas patologías para poder efectuar un rápido diagnóstico y tratamiento, dado que el pronóstico a largo plazo puede depender de una rápida intervención diagnostica. |
| Abstract
The embryology of the formation of the hepatobiliary system is reviewed and the clinical, diagnostic and therapeutic aspects of the most frequent pathologies of neonatal presentation such as bile duct atresia, choledochal cyst and intrahepatic bile duct paucity are also described. The typical presentation involves neonatal cholestasis with the classic triad of jaundice, acolia and hepatomegaly. It is important to be familiar with these pathologies so as to make a rapid diagnosis and treatment since long-term prognosis may depend on a prompt diagnostic intervention. |
Palabras clave: Embriología hepatobiliar; Colestasis neonatal.
Key words: Hepatobiliary embryology; Neonatal cholestasis.
Pediatr Integral 2020; XXIV(1): 56 – 59
Recuerdo anatómico patológico del sistema hepatobiliar
Introducción
Muchas enfermedades tienen su origen en un defecto en la fase de desarrollo embrionario, dando lugar a una disfunción de la parte afecta. Nos referiremos solamente a aquellas patologías más frecuentes, que tienen su presentación en la época de recién nacido y que son consecuencia de una alteración anatómica del sistema hepatobiliar. Se obviarán aquellas patologías como: enfermedades metabólicas, anomalías en la síntesis o transporte de las sales biliares, infecciones u otras noxas patológicas intrauterinas, que puedan conllevar una disfunción en el funcionamiento del sistema hepatobiliar embriológicamente sano en su inicio y que hacen su aparición en periodo neonatal, y entran dentro del diagnóstico diferencial de colestasis neonatal.
Recuerdo embriológico
En el embrión, el hígado se desarrolla como una evaginación endodérmica de la pared del intestino anterior para formar el divertículo hepático. El divertículo prolifera y da origen a los hepatocitos, que se organizan en láminas o trabéculas que formarán el parénquima hepático. El pedículo original del divertículo hepático se convertirá en el colédoco. De este conducto saldrá un brote que formará el divertículo cístico y que dará origen a la vesícula biliar y al conducto cístico(1) (Fig. 1).
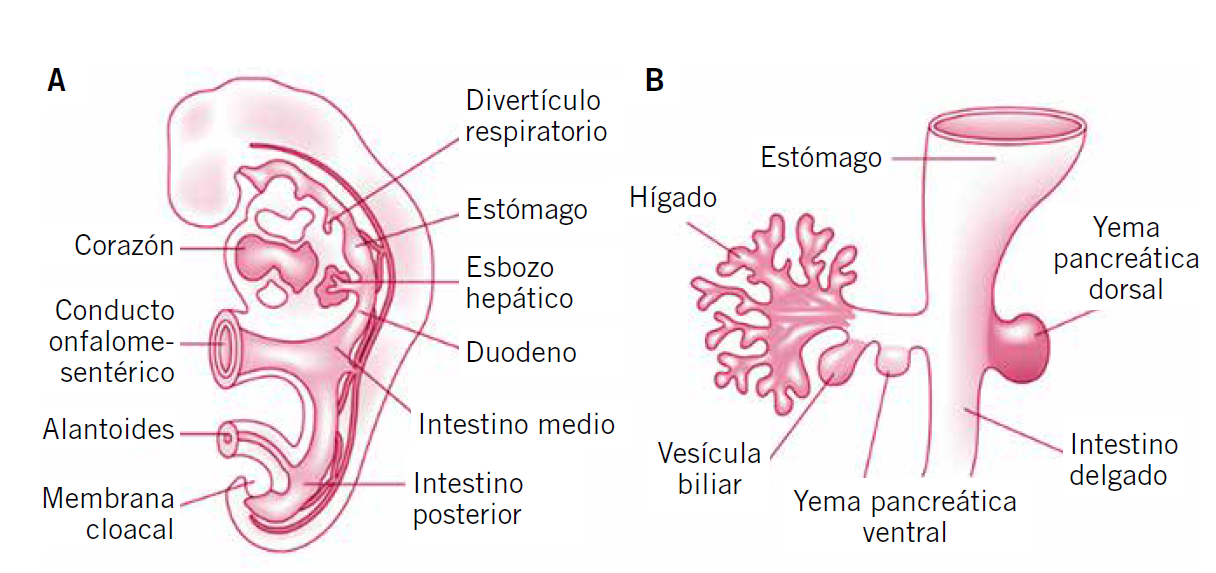
Figura 1. Desarrollo embriológico del sistema hepatobiliar.
Recuerdo anatómico
El hígado es la víscera más grande del cuerpo, se sitúa en el hipocondrio derecho en su mayor parte y en el epigastrio, y corresponde al 2,5% del peso corporal. A nivel estructural microscópico, el parénquima hepático está formado por los lobulillos hepáticos que son la unidad funcional básica
(Fig. 2).
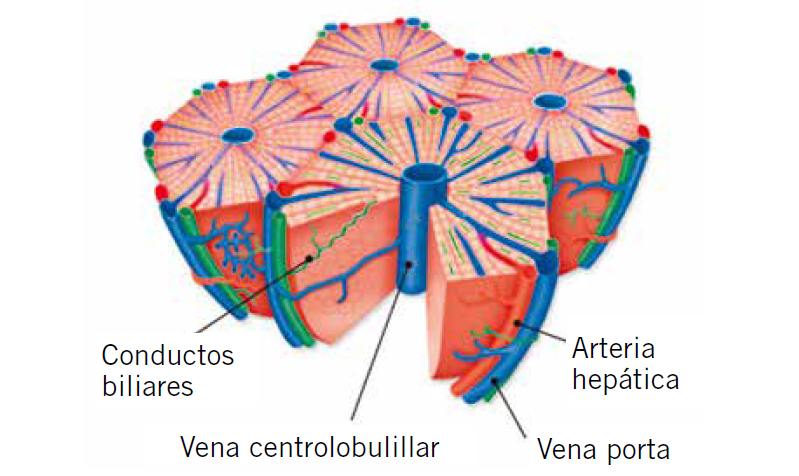
Figura 2. Estructura microscópica del lobulillo hepático.
Cada lobulillo se compone de trabéculas de hepatocitos anastomosados que se alejan de la vena centrolobulillar en disposición radial. Además, cada lobulillo consta de conductos biliares que discurren por los tabiques fibrosos que separan cada lobulillo hepático. Los tabiques separadores también llevan vénulas portales y arteriolas hepáticas. Las vénulas portales reciben sangre del tubo digestivo y, desde estas vénulas, la sangre se dirige a los sinusoides venosos y, posteriormente, a la vena central centrolobulilllar. La vena central del lobulillo desembocará en las venas hepáticas y, posteriormente, en la vena cava inferior. Los sinusoides venosos están revestidos de unas células grandes denominadas células de Kupffer, que son macrófagos que fagocitan bacterias y otros cuerpos extraños provenientes de la sangre(2).
A nivel anatómico (Fig. 3), podemos distinguir la cara diafragmática o anterior, superior y posterior, y la cara visceral en la parte inferior, que está recubierta de peritoneo, excepto en la fosa de la vesícula biliar y el hilio hepático.
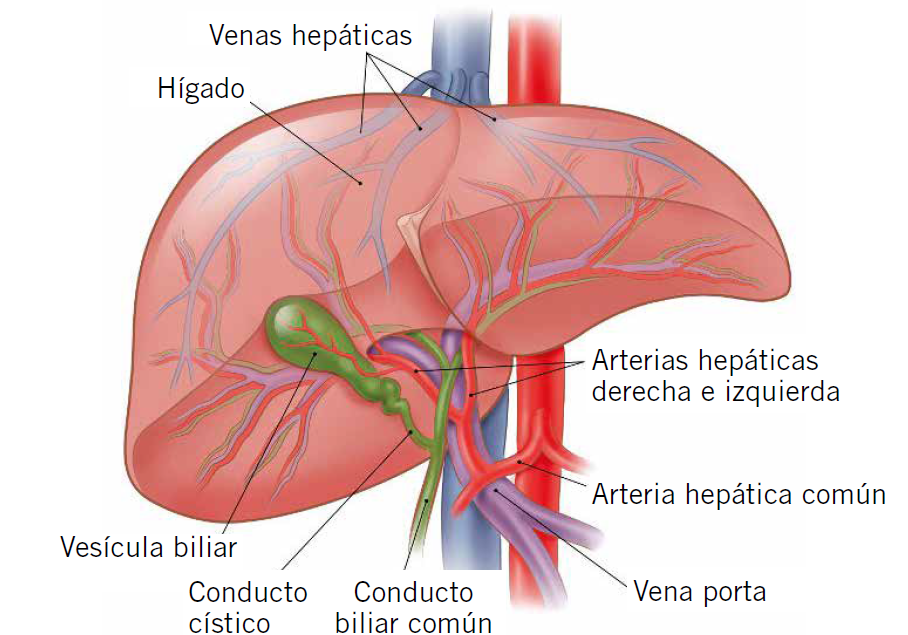
Figura 3. Recuerdo anatómico del sistema hepatobiliar.
El hígado está dividido por: la vesícula biliar, la vena cava y las arterias hepáticas en dos lóbulos grandes, derecho e izquierdo. El hilio hepático es el punto de entrada de las arterias hepáticas derecha e izquierda, ramas de la arteria hepática común originada en el tronco celíaco, y la vena porta. Así mismo, es el punto de salida de los conductos hepáticos.
Los conductos biliares son un sistema de vasos para el paso de la bilis desde el hígado hasta la porción descendente del duodeno. Las ramas más pequeñas del sistema son los canalículos biliares, hacia los cuales los hepatocitos secretan la bilis. Estos conductos empiezan en el parénquima hepático y continúan hasta la formación del conducto hepático izquierdo y derecho. Estos se unen formando el conducto hepático común, que va junto a la arteria hepática y la vena porta, formando el hilio hepático. En el descenso, el conducto hepático se une al conducto cístico (que proviene de la vesícula biliar) y forman el colédoco. El colédoco se unirá al conducto pancreático para penetrar en la porción descendente del duodeno o papila duodenal mayor.
La vesícula biliar es un saco con forma de pera, situado en la cara visceral del lóbulo hepático derecho. Está formada por el fondo, el cuerpo que se apoya en el colon transverso o parte superior del duodeno, y el cuello. La irrigación proviene de la arteria cística, rama de la arteria hepática derecha. La vesícula biliar se encarga de concentrar y almacenar la bilis que viene del hígado(3).
Hepatopatías por trastorno hepatobiliar intrínseco, por defecto del desarrollo embriológico
Son las hepatopatías colestaticas más frecuentes en la edad infantil. El término colestasis define aquella situación en la que existe una alteración del flujo biliar, con la consiguiente retención y paso a sangre de componentes de la bilis (bilirrubina directa, sales biliares, colesterol, etc.), y que condiciona un cuadro clínico característico, con: ictericia, aparición de bilirrubina en orina (coluria), decoloración parcial o completa de las deposiciones (hipo o acolia) y prurito; así como, un cuadro bioquímico con: aumento de bilirrubina directa, GGT, fosfatasa alcalina y colesterol y, debido a la citolisis biliar, aumento de las transaminasas. Tres son las entidades más frecuentes: la atresia de vías biliares extrahepática, los quistes del colédoco y la paucidad de vías biliares intrahepática.
Atresia de las vías biliares extrahepática
Es la causa más frecuente de colestasis crónica y de trasplante hepático en la infancia, con una incidencia media de 1/10.000 RN vivos. Se reconocen dos formas clínicas: la embrionaria o sindrómica (10% de los casos), que se puede asociar a otras malformaciones (polisplenia, situs inverso, etc.), y la forma perinatal o adquirida (90% de los casos), con mecanismos patogénicos probablemente diferentes; con defecto en la morfogénesis en la primera, y una posible agresión viral o toxica perinatal con respuesta inmune en la segunda(4).
En esta última, existe un proceso inflamatorio progresivo que conduce a la obliteración de la vía biliar extrahepática, así como a la lesión del parénquima hepático (inflamación y fibrosis) y de la vía biliar intrahepática. Aunque no se puede considerar un defecto de la embriogénesis, se suele incorporar en el contexto de “atresia biliar extrahepática”.
El cuadro clínico es muy característico, con un recién nacido a término, de peso y aspecto normal, que inicia ictericia con hipocolia entre las 2 y las 6 semanas de vida, con buen estado general, hepatomegalia firme, y posterior esplenomegalia. Analítica con signos de colestasis (aumento de bilirrubina total y directa, GGT muy elevada), elevación moderada de transaminasas y coagulación normal.
Los hallazgos ecográficos dependen del tipo y de la gravedad de la atresia de vías biliares(5). El tamaño del hígado puede estar normal o aumentado. Los conductos biliares intrahepáticos normalmente no están dilatados. Puede visualizarse un remanente de la vía biliar extrahepática a la altura del hilio hepático, dependiendo del tipo de atresia biliar. Este aparece como una estructura ecogénica triangular o tubular, justo por encima de la bifurcación de la porta. Este hallazgo se llama el “cordón triangular” y se correlaciona con el tejido fibroso que se encuentra en hilio en el examen histológico. Se observa una hipoplasia de la vesícula y la vía biliar(6).
Otras pruebas que nos pueden ayudar en el diagnóstico, son: la realización de una gammagrafía hepática tras la administración de fenobarbital, donde se observará una ausencia de excreción intestinal. En la biopsia, encontraremos hallazgos compatibles con: patrón de colestasis, proliferación ductal y fibrosis portal. La prueba confirmatoria será la laparotomía o laparoscopia exploradora, con la realización directa de una colangiografía intraoperatoria.
El tratamiento es quirúrgico, con realización de una porto-entero-anastomosis (Kasai 1) de cara al restablecimiento del flujo biliar y consiguiente mejora del pronóstico. Son fundamentales, el diagnóstico y tratamiento precoces antes de los dos meses de vida. Sin tratamiento, la mortalidad es del 100% antes de los 3 años de vida, por desarrollo de cirrosis biliar e insuficiencia hepática. Tras la cirugía, un 30% no restablecerán flujo biliar y otro 20% lo harán de forma parcial, precisando todos ellos un trasplante hepático en los meses siguientes. Del 50% restante que restablece el flujo biliar tras la cirugía, un 70% de ellos precisará, a largo plazo, trasplante hepático por evolución cirrógena, ya que la lesión del parénquima persiste a pesar del buen resultado quirúrgico. La supervivencia a los dos, cuatro y diez años de vida, sin necesidad de trasplante, se sitúa alrededor del 55%, 45 y 35%, respectivamente. Se han referido como factores predictivos de mala evolución post-Kasai: la forma embrionaria de la enfermedad, la existencia de fibrosis severa, la edad superior a los dos meses en el momento de la cirugía o la falta de experiencia, tanto quirúrgica como en el manejo postoperatorio del centro. Posteriormente al trasplante, la supervivencia global es de hasta el 90% a los 10 años de vida(7).
Quiste de colédoco
La etiología es desconocida, pero la teoría más aceptada es la unión anómala de los conductos pancreáticos y biliares fuera del duodeno. Esto conlleva a un reflujo del jugo pancreático al árbol biliar, que condiciona una inflamación y deterioro del conducto, que daría lugar a la dilatación biliar. La incidencia es de 1/13.000 a 1/2.000.000 de recién nacidos vivos, con predominio en poblaciones asiáticas. Se pueden clasificar en 5 tipos, según su localización y forma (Tabla I).

Normalmente, se diagnostica en la infancia, la tríada clásica de: ictericia, dolor abdominal y masa abdominal palpable en hipocondrio derecho. Muchas veces, el dolor abdominal recurrente es el síntoma más frecuente. También, se puede asociar: colangitis, pancreatitis e hipertensión portal secundarias. Además, por la estasis biliar, podemos encontrar signos de colecistitis aguda o litiasis en la vesícula biliar. La ruptura quística es poco frecuente, ocurre solamente en neonatos y lactantes. En niños más mayores o adultos, puede no dar síntomas. A veces, se asocia con dolor abdominal e ictericia intermitente. A diferencia de cuando son más pequeños, la palpación de la masa abdominal es infrecuente.
En la enfermedad de Caroli (quistes de colédoco tipo V), los quistes intrahepáticos pueden ser únicos o múltiples, convergen hacia la porta y representan una dilatación sacular de los conductos biliares. Los vasos portales están rodeados por conductos dilatados de forma parcial o completa y, además, puede haber dilatación de la vía biliar común. Se puede asociar a: fibrosis hepática, hipertensión portal y a la enfermedad renal poliquística(8).
En la ecografía, los quistes de colédoco de los tipos I, II y III aparecen como un quiste simple en la región del conducto biliar común, que puede estar separado de la vesícula biliar. Para el diagnóstico de quiste de colédoco, se debe demostrar la comunicación con el sistema ductal biliar. Los conductos intrahepáticos pueden estar dilatados o no. Se debe explorar el páncreas y el conducto pancreático, para descartar pancreatitis o dilatación ductal. En la enfermedad de Caroli, los quistes intrahepáticos periportales pueden acompañarse de la dilatación de la vía biliar extrahepática(6).
Entre los exámenes de laboratorio, podemos encontrar: alteración de las enzimas hepáticas junto con aumento de la amilasa, lipasa y leucocitosis. Si hay alteración en la coagulación y en la función renal, serán indicadores de gravedad.
El tratamiento dependerá del tipo de quiste. Generalmente es quirúrgico; en el quiste tipo I, se recomienda la cirugía con resección. La cirugía más utilizada es la quistectomía con reconstrucción de la vía biliar mediante hepatoyeyunostomía de Y de Roux. En los quistes tipo III, el tratamiento de elección es la esfinterotomía endoscópica mediante colangiopancreatografía retrógrada endoscópica (CPRE). En los quistes tipo V o enfermedad de Caroli con afectación unilobular, el tratamiento más efectivo es la hepatectomía parcial del lóbulo afectado. Si hay afectación difusa, el tratamiento inicial deberá ser con ácido ursodesoxicólico y sales biliares quelantes; pero si se produce cirrosis biliar secundaria, será necesario el trasplante hepático.
Como otras complicaciones, encontramos: litiasis biliar, litiasis hepática, colangitis, pancreatitis aguda y crónica, cirrosis biliar y complicaciones derivadas de la hipertensión portal.
Paucidad de las vías biliares sindrómica: síndrome de Alagille
Se caracteriza por la asociación de una colestasis con: escasez de conductos biliares intrahepáticos, alteraciones cardíacas (la más frecuente, estenosis periférica de la arteria pulmonar), alteraciones vertebrales (vértebras en mariposa), oculares (embriotoxon posterior) y facies peculiar con hipertelorismo, abombamiento frontal y mentón prominente. También presentan: anomalías renales (quistes, hipoplasisa…), dentarias, vasculares o disminución de la capacidad intelectual con menor frecuencia(9-10). Como mecanismo patogénico, se considera una vasculopatía secundaria a los genes JAG 1 (95%) o NOTCH2 (1%). La mitad de los casos son hereditarios, con una herencia autosómica dominante con penetrancia prácticamente completa (96%), pero con gran variabilidad de expresión clínica. Es muy frecuente la repetición del síndrome en los hermanos de un niño afecto. La otra mitad son mutaciones de novo(9).
En el 80% de los pacientes, la colestasis es de inicio neonatal o en la época del lactante pequeño, y se acompaña de: ictericia, hipocolia, hepatomegalia y retraso ponderal, además de prurito intenso y xantomas. El 20-30% restante, presentarán signos de hepatopatía pasado el periodo de lactante, asociarán prurito leve y subictericia fluctuante. En la analítica presentan: hiperbilirrubinemia, elevación de los ácidos biliares, hipercolesterolemia grave, elevación de la GGT y de las transaminasas. La importante colestasis da lugar a déficit severo de las vitaminas liposolubles. El déficit de vitaminas puede llevar a: raquitismo, fracturas de repetición, retinopatía pigmentaria, malnutrición y sangrado, y malnutrición severa. Aún con tratamiento médico, el crecimiento es inferior a la normalidad, son frecuentes la osteoporosis y las fracturas patológicas.
El diagnóstico del síndrome se basa fundamentalmente en criterios clínicos. El diagnóstico de certeza se realiza con la biopsia hepática, cuando se constata la escasez de conductos biliares interlobulares; además debe asociar, al menos, tres de los siguientes rasgos: colestasis, cardiopatía, embriotoxon posterior, vértebras en mariposa, cara peculiar, enfermedad renal o antecedente familiar. En niños menores de seis meses, en los que no puede objetivarse la escasez de conductos biliares en la biopsia, la asociación de 3 o 4 criterios es suficiente para el diagnóstico. El diagnóstico genético puede encontrar mutaciones hasta en el 95% de individuos con criterios clínicos, por lo que en pacientes que no cumplen todos los criterios clínicos, es necesario. También el diagnóstico genético será necesario para familiares del sujeto sin criterios clínicos.
El síndrome de Alagille no tiene tratamiento específico, es el común para todo cuadro colestásico. El tratamiento con: fenobarbital, ácido ursodesoxicólico y la suplementación con vitaminas liposolubles, mejora la evolución de la enfermedad, disminuyendo la intensidad de los síntomas. Es de vital importancia, optimizar el estado nutricional de estos pacientes para prevenir las complicaciones asociadas. Su evolución natural es el desarrollo, en un 50% de los casos, de una colestasis grave y, posteriormente fibrosis, que requerirán de un trasplante hepático antes de la vida adulta. El pronóstico de supervivencia a los 10 años es de un 45% de los que habían debutado en periodo neonatal, y un 79% de los que tenían inicio más tardío(7).
Bibliografía
1. Guyton AC, Hall JE. Tratado de Fisiología médica. Edición 8ª. Interamericana-McGraw-Hill. Madrid.
2. Ross MH. Histología Texto y Atlas, 2ª edición. Ed. Médica Panamericana. 2005.
3. Williams PL. Anatomía de Gray. 38ª edición. Harcourt Brace de España S.A. Madrid.
4. Feldman AG, Mack CL. Biliary Atresia: Clinical Lessons Learned. J Pediatr Gastroenterol Nutr. 2015; 61: 167-75.
5. Guideline for the Evaluation of Cholestatic Jaundice in Infants: Joint Recommendations of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition and the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition. J Pediatr Gastroenterol Nutr. 2017; 64: 154-68.
6. Osiniri Kippes I. La ecografía clínica permite visualizar la causa de la ictericia patológica en lactantes y niños. Pediatr Integral. 2015; XIX(3): 224.e1–224.e10.
7. de la Vega A, Frauca Remacha E. Síndrome colestático. Actitud diagnóstico-terapéutica. Pediatría Integral. 2015; XIX(3): 168-78.
8. Soares KC, Goldstein SD, Ghaseb MA, Kamel I, Hackam DJ, Pawlik TM. Pediatric choledochal cysts: diagnosis and current management.. Pediatr Surg Int. 2017; 3: 637-50.
9. Alagille D, Odievre M, Gautier M. Hepatic ductular hypoplasia associated with characteristic facies, vertebral malformations, retarded physical, mental, and sexual development, and cardiac murmur. J Pediatr. 1975; 86: 63-71.
10. Squires JE, McKiernan P. Molecular Mechanisms in Pediatric Cholestasis. Gastroenterol Clin North Am. 2018; 47: 921-37.
