 |
| Temas de FC |
L. Hierro Llanillo, G. Muñoz Bartolo
Servicio de Hepatología Pediátrica. Hospital Infantil Universitario La Paz, Madrid
| Resumen
Las enfermedades primarias del hígado son patologías raras. En las hepatopatías colestáticas es necesario: soporte nutricional, vitaminas, profilaxis y tratamiento de infecciones. Las colestasis iniciadas en el periodo neonatal, en una elevada proporción, requieren trasplante hepático en edad infantil. La hepatitis autoinmune y la enfermedad de Wilson son el prototipo de enfermedad hepática con tratamiento médico eficaz, para lo que requieren apoyo y vigilancia para el buen cumplimiento. La elevada supervivencia del paciente y del injerto tras el trasplante hepático han motivado un interés creciente por el pronóstico a largo plazo y la calidad de vida. Los esfuerzos actuales van dirigidos a minimizar las complicaciones a largo plazo, manteniendo la función del injerto normal. Muchas de las complicaciones están relacionadas con la medicación inmunosupresora que reciben estos pacientes. El conocimiento de este tratamiento y de los posibles problemas después del trasplante, ayuda a su correcto manejo. Es importante la colaboración multidisciplinar. |
| Abstract
Primary diseases of the liver are rare entities. Cholestatic children need nutritional support, vitamins, prevention and treatment of infections. A high proportion of the cholestatic diseases of neonatal onset will require liver transplantation at an early age. Autoimmune hepatitis and Wilson are the prototypes of liver diseases with an effective treatment, for which they need continuous support for compliance. |
Palabras clave: Colestasis neonatal; Hepatitis autoinmune; Enfermedad de Wilson; Trasplante hepático; Tacrolimus..
Key words: Infant cholestasis; Autoimmune hepatitis; Wilson´s disease; Liver transplantation; Tacrolimus.
Pediatr Integral 2020; XXIV(1): 47 –55
Seguimiento del niño con hepatopatía crónica y trasplante
Seguimiento en niños con hepatopatía crónica
Las hepatopatías crónicas son un gran número de entidades. En una sociedad pueden predominar enfermedades adquiridas, como las hepatitis infecciosas y la hepatopatía relacionada con obesidad. En este capítulo solo se considerarán las hepatopatías primarias.
La enfermedad más frecuente es la atresia biliar, que afecta a 1 de cada 18.000 recién nacidos. El síndrome de Alagille es la colestasis intrahepática más frecuente (1 por 30.000-70.000). Las colestasis intrahepáticas familiares afectan a 1 por 200.000 niños. La enfermedad de Wilson tiene una incidencia de 1 por 30.000. La hepatitis autoinmune ocurre en 1,6-3 por 100.000.
Atresia biliar
La causa es desconocida. Hay una inflamación hepática y biliar perinatal que causa fibrosis de la vía biliar extrahepática con obstrucción completa. Los niños tienen: ictericia, coluria y deposiciones de color blanquecino o amarillo muy pálido (acolia), sin afectar a su estado general en el primer mes de vida. Después habrá esplenomegalia y hepatomegalia de consistencia aumentada.
Diagnóstico y tratamiento quirúrgico
Todos los recién nacidos con ictericia persistente a las 2-3 semanas, deben ser evaluados con analítica. Las heces deben ser visualizadas.
Dado que el pronóstico está influido por la edad de detección, es importante recordar que todo niño con ictericia persistente más de 2-3 semanas, debe ser estudiado (una hepatopatía sería evidenciada por aumento de bilirrubina directa mayor de 1 mg/dl) y es siempre necesaria la observación directa del color de las heces, la descripción hecha por la familia no es fiable(1). La detección precoz permite derivar al paciente a un centro de referencia, para diagnóstico diferencial y tratamiento.
La combinación de peso normal al nacimiento, acolia, GGT elevada (>300 UI/L) y ausencia de insuficiencia hepática (coagulación normal o normalizada con vitamina K), son predictivos (90%) de atresia biliar(2).
La confirmación se realiza con colangiografía en laparoscopia o laparotomía, Se realizará una porto-enterostomía (técnica de Kasai) que consiste en la disección y extirpación del remanente fibroso biliar a nivel del hilio hepático, sección a nivel del hilio (placa ductal) y conexión a esa zona de una asa yeyunal (Y de Roux), que permita el drenaje de bilis desde los pequeños conductos biliares de la placa ductal. Esta cirugía pretende solucionar el componente obstructivo que existe en la enfermedad. No obstante, la enfermedad afecta también al hígado y ello determina la evolución.
La recuperación del flujo biliar tras la porto-enteroanastomosis de Kasai, se asocia a una supervivencia sin trasplante durante los siguientes años.
La edad inferior a 45 días se acompaña de mejor tasa de recuperación de flujo biliar, comparado con niños operados a edad superior(3)
Plan de seguimiento de un paciente con atresia biliar
En los primeros 3 meses postKasai, se observa el efecto de la cirugía. El óptimo es la ausencia de ictericia y bilirrubina total <2 mg/dl.
En algunos niños, la desaparición de la ictericia sucede en el primer mes, pero debido a que la cirugía acompaña un riesgo de infección del hígado y hay diferentes grados de afectación previa del hígado, es necesario dar un margen de 3 meses para una conclusión de eficacia. El estado óptimo, ausencia de ictericia y bilirrubina sérica total <2 mg/dl, se obtiene en un 40-50% de los operados. Otros niños tienen una recuperación parcial, con heces coloreadas, pero bilirrubina elevada. La persistencia de ictericia franca con bilirrubina total >6 mg/dl identifica al grupo que deben ser cuidados especialmente, por riesgo de progresión a muy corto plazo, a complicaciones de hipertensión portal e insuficiencia hepática(4). En ausencia de trasplante, el fallecimiento ocurrirá entre los 9 y 18 meses.
El tratamiento postKasai con esteroide oral 1-6 meses o con inmunoglobulina iv se ha demostrado ineficaz. Actualmente, está en ensayo la administración de acetilcisteina iv(5).
A partir del 3er mes postKasai, debe ser indicado trasplante hepático en los pacientes que no han restablecido flujo biliar.
Los niños con restablecimiento parcial siguen un control estrecho, y la observación de: infecciones repetidas, dilataciones biliares focales, malnutrición o progresión de signos de hipertensión portal, son los criterios para indicar trasplante. Los niños con buen restablecimiento de flujo biliar son heterogéneos en sus datos clínicos (colangitis, progresión de esplenomegalia), pero aproximadamente la mitad de ellos son supervivientes a largo plazo, con leves signos de hipertensión portal y casi normalidad bioquímica.
Tratamiento médico
Incluye un control estrecho del estado nutricional, indicando una fórmula láctea (exclusiva o complementaria de lactancia materna) con grasa en forma de triglicéridos de cadena media (MCT), suplementos calóricos [dextrinomaltosa (DMT) antes de los 3 meses, después cereales] y no se modifica la diversificación de alimentos, Puede ser necesario aportar nutrientes por SNG (sonda nasogástrica). Todos requieren suplementos de vitaminas liposolubles (Tabla I).
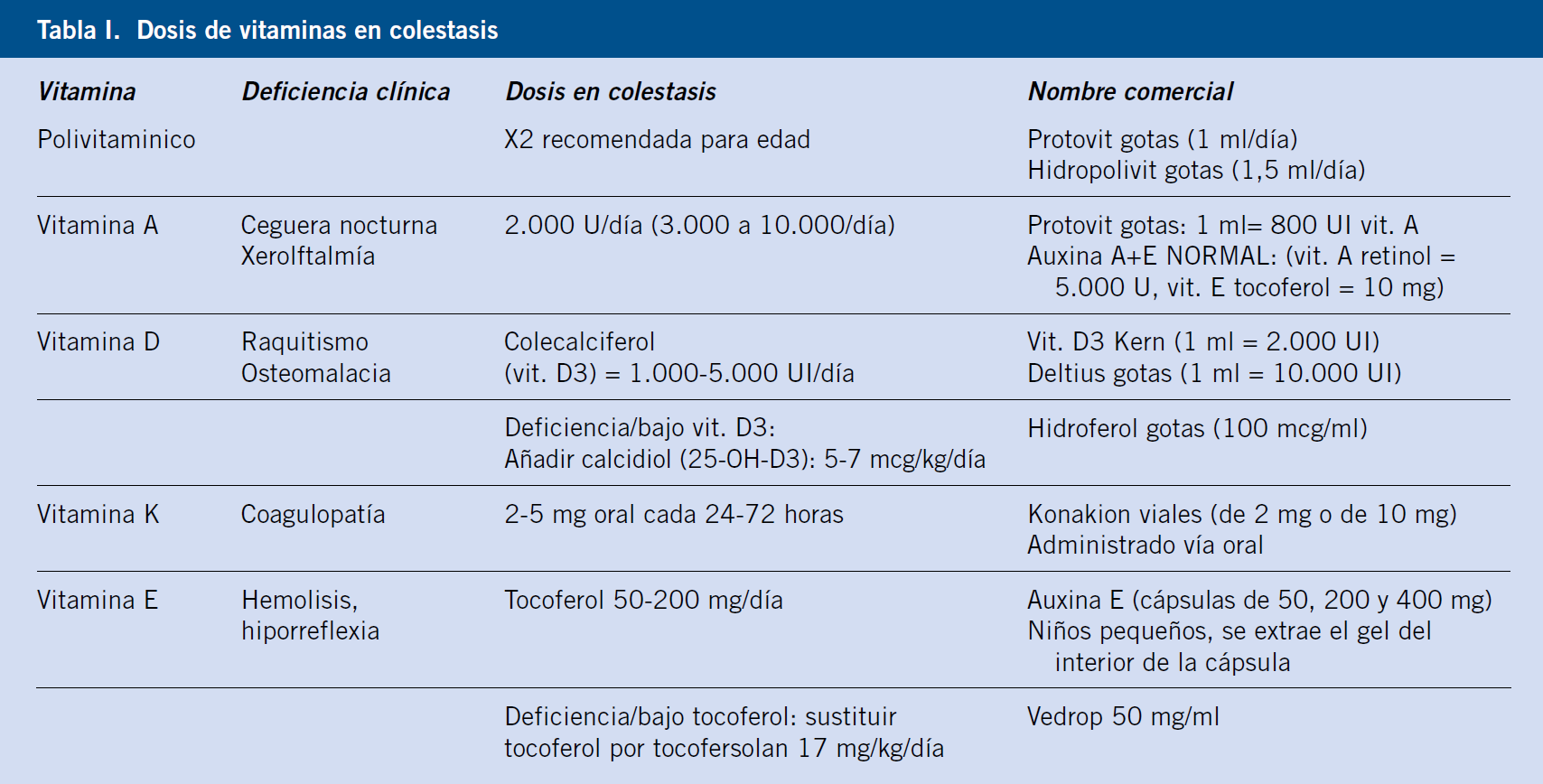
Las pautas de cuidado incluyen la administración de ursodeoxicólico (15 mg/kg/día) para favorecer el flujo biliar y como protector hepático, pero esta medicación debe ser retirada si no se ha logrado restablecer secreción biliar.
Los antibióticos orales (amoxicilina o cotrimoxazol), a dosis de tratamiento, son administrados 3 a 12 meses postKasai, como prevención de colangitis.
Complicaciones
Un empeoramiento de la hepatopatía asociado a fiebre, justifica la consulta o derivación al centro de referencia.
La aparición de colangitis se sospecha por fiebre y empeoramiento de valores de bilirrubina o de AST/ALT/GGT. En un contexto de infección ORL o digestiva, podría ser manejado inicialmente con conversión a amoxi-clavulánico o cefuroxima axetilo, o cefixima, pues cualquier infección puede aumentar la disfunción hepática. En caso de progresión o sospecha de infección primaria en hígado, el paciente debe ser hospitalizado y tratado con antibióticos por vía intravenosa (cefotaxima o piperacilina tazobactam, como primera indicación).
La hipertensión portal causa ascitis en niños, con ausencia de restablecimiento de flujo biliar, a partir de los 6 meses de edad. Acompaña hipoalbuminemia por insuficiente síntesis. Se indica espironolactona oral (4 mg/kg/día). Si persiste o progresa, se asocia albúmina iv y diuréticos más potentes (furosemida) y terlipresina.
Hay riesgo de hemorragia digestiva por medicación gastroerosiva, que debe ser siempre evitada (para fiebre o dolor, se recomienda metamizol).
En estadios de hepatopatía avanzada, se asocia ranitidina oral. La profilaxis primaria de hemorragia (ligadura de varices o propranolol) no está indicada de forma general, ya que en niños pequeños, no es posible, por el calibre de los endoscopios, efectuar ligadura de varices, y la medicación con propranolol favorecería hipotensión en sangrado. Sin embargo, algunos centros realizan profilaxis primaria(6). Si se produce hemorragia, está indicado realizar profilaxis secundaria [esclerosis de varices y colocación de TIPs (transjugular intrahepatic portosystemic shunt)] y aportar hemoderivados (plasma, plaquetas), según la analítica del paciente. Los niños con atresia que sobreviven a largo plazo sin ictericia, pero con signos de hipertensión portal, pueden beneficiarse de profilaxis primaria de hemorragia (ligadura o propranolol), tras una valoración endoscópica del riesgo. El trasplante hepático es la indicación terapéutica para la hipertensión portal severa de los niños con atresia que sobreviven a largo plazo.
Colestasis intrahepática (síndrome de Alagille -SA-) y colestasis intrahepática familiar progresiva (CIFP)
La hepatopatía del síndrome de Alagille es debida a un desarrollo embrionario alterado en los conductos biliares intrahepáticos. Las colestasis intrahepáticas familiares progresivas son debidas a un defecto genético en el proceso de secreción canalicular (transportador, membrana, unión intercelular).
El síndrome de Alagille (mutación en 1 alelo, genes JAG1 o NOTCH2) causa colestasis y asocia: peso bajo al nacer, características faciales, soplo sistólico por hipoplasia de arterias pulmonares o alteraciones renales (hipoplasia). Presentan AST/ALT y GGT elevadas, y una cifra de colesterol alta, a veces, con xantomas (Fig. 1).

Figura 1. Xantomas en síndrome de Alagille.
Causa prurito intenso y refractario, desde los 4-7 meses de vida(7).
Las CIFP son entidades raras de colestasis con prurito, en las que existe un defecto genético (autosómico recesivo). La forma más frecuente afecta al transportador de ácidos biliares (BSEP) en la membrana canalicular(8).
Pronóstico
La intensidad de los síntomas colestáticos, prurito y mal crecimiento determinan la indicación del trasplante hepático (a edad 1-5 años en su mayoría), antes de que en el curso natural ocurra cirrosis y sus complicaciones. Un 70% de síndrome de Alagille y un 80% de CIFP lo requieren en edad pediátrica,
Tratamiento médico
El tratamiento de sostén es esencial para evitar deficiencias de vitaminas liposolubles, requieren generalmente dosis elevadas y vitamina E especial (tocofersolan)(9). El apoyo nutricional, semejante al descrito en atresia biliar, es imprescindible. No requieren profilaxis de colangitis, pero se benefician del uso de antibióticos en las infecciones intercurrentes, que causan aumento del prurito. Los niños con síndrome de Alagille tienen predisposición a otitis recurrentes.
La medicación empleada para la colestasis y el prurito incluye: ácido ursodeoxicólico (15 mg/kg/día), fenobarbital (3 mg/kg/día) y dosis a demanda de antihistamínico (Tabla II).
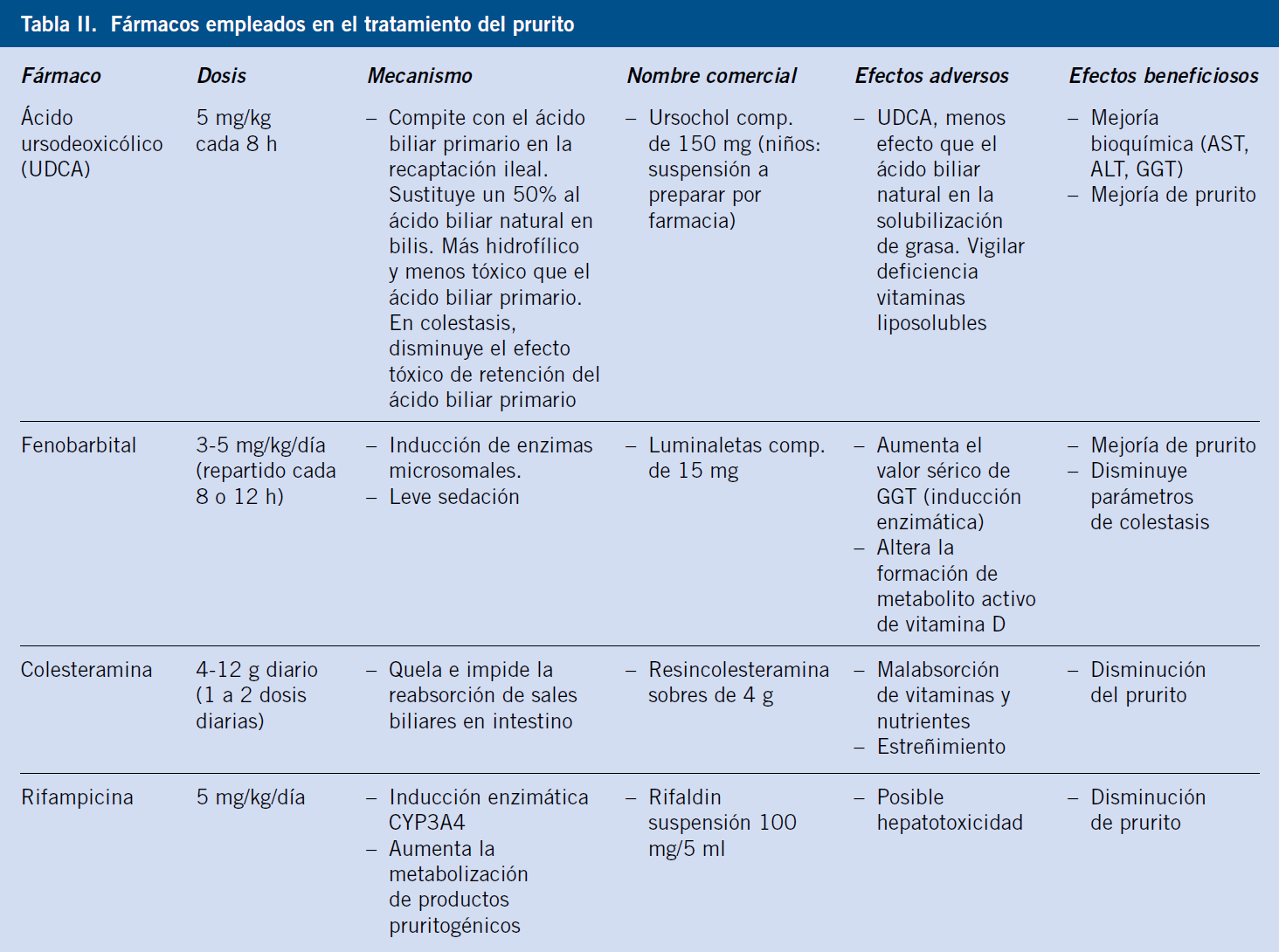
Los fibratos, resincolesteramina, rifampicina y sertralina pueden ser usados, pero conllevan riesgos(10). Hay fármacos en investigación (maralixibat) inhibidores de la reabsorción de ácidos biliares en el íleon distal(11). En CIFP por defecto de FIC1 y SA, puede desaparecer el prurito con pérdida neta de bilis, realizando una derivación biliar externa parcial con interposición de un segmento de intestino entre la vesícula y el exterior (estoma en abdomen).
Hepatitis autoinmune (HAI) y enfermedad de Wilson (EW)
La hepatitis autoinmune y la enfermedad de Wilson son susceptibles de tratamiento eficaz, que debe ser continuado en el tiempo. El diagnóstico en niños o adolescentes con aminotransferasas elevadas, facilita su recuperación completa.
Presentación clínica
Muchos (60% HAI y 85% EW) son diagnosticados en nuestro medio, con un correcto proceso de estudio (determinación de autoanticuerpos y de ceruloplasmina) a partir de la detección de alteración de AST/ALT en un chequeo por síntomas no específicos. La detección en estadio temprano, permite la respuesta al tratamiento específico de cada enfermedad.
Un 40% de HAI y un 15% de EW se diagnostican sintomáticos, con ictericia y grave alteración funcional. La mitad de ellos serán reversibles con tratamiento.
Tratamiento
En HAI, el tratamiento de elección son: esteroides (inicial 2 mg/kg/día máximo 60 mg/día) y azatioprina (1 mg/kg/día)(12). En intolerancia a azatioprina (citopenia o vómitos) o falta de respuesta, se indica cambio a micofenolato o tacrolimus(13). Se define remisión a la obtención de bioquímica hepática e IgG normales. La guía para la medicación continuada es el mantenimiento de remisión con los mínimos efectos adversos. El descenso de esteroide debe ser gradual e individualizado, comprobando el mantenimiento de la remisión.
La educación del paciente con HAI y su familia es necesaria para evitar la excesiva ganancia de peso inicial, precisan vigilancia de la ingesta y fomento del ejercicio físico.
Los niños con EW tardan aproximadamente un año en normalizar las alteraciones analíticas hepáticas bajo tratamiento, después deben continuar normales si lo cumplen adecuadamente. La elección es penicilamina o acetato de zinc, según la intensidad de la alteración inicial(14). La dificultad del tratamiento es la necesidad de ayuno, 1 hora antes y 1 hora después de tomar el medicamento. La dieta se restringe indefinidamente en los alimentos con alto contenido de cobre (chocolate, frutos secos, setas y foie).
Seguimiento del trasplante hepático pediátrico
El desarrollo de los fármacos inmunosupresores, el avance de las técnicas quirúrgicas y de los cuidados médicos ha permitido alcanzar una elevada supervivencia, tanto del paciente como del injerto tras el trasplante hepático, en sus diferentes indicaciones (Fig. 2).
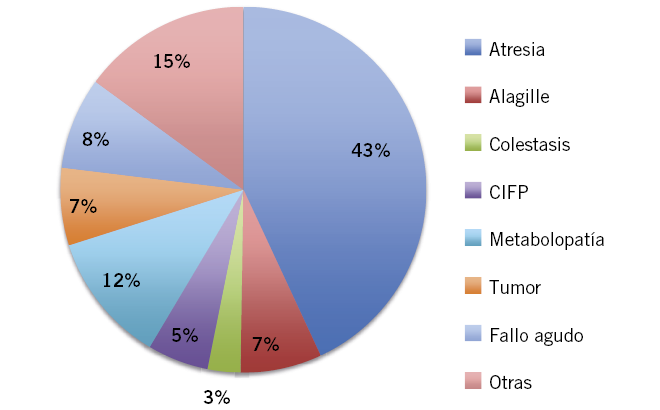
Figura 2. Indicación de trasplante hepático en niños. Hospital infantil La Paz, Madrid. Período 1990 a 2016, n = 555, 50% de trasplantes se realizan en < 2 años.
La supervivencia del paciente es superior al 90% al año y al 85% a los 10 años(15) Los esfuerzos actuales van dirigidos, no solo a preservar la función del injerto y la supervivencia del paciente, sino a minimizar las complicaciones a largo plazo, algunas de ellas derivadas del uso de inmunosupresores.
Los excelentes resultados (Fig. 3) han motivado un creciente interés por la calidad de vida.

Figura 3. Supervivencia del paciente. Trasplante hepático Hospital La Paz. Madrid. Período 2000-2018.
La colaboración conjunta con los pediatras de Atención Primaria es muy importante. A continuación, se exponen algunas de las complicaciones que pueden ocurrir en el seguimiento y recomendaciones de interés. El esquema de revisiones se resume en las tablas III y IV.


Aspectos quirúrgicos relevantes. Profilaxis de trombosis vascular. Complicaciones biliares
y vasculares a largo plazo
El trasplante hepático pediátrico tiene unas peculiaridades técnicas que lo diferencian del de adultos(16).
El injerto puede proceder de donante vivo o cadáver (entero o reducido). La anastomosis biliar habitual en niños es la hepaticoyeyunostomía en Y Roux. Las anastomosis vasculares son difíciles y exigen una gran experiencia del equipo quirúrgico. Las trombosis de la arteria hepática y de la vena porta, poco frecuentes, pueden requerir retrasplante hepático urgente.
• Complicaciones vasculares a largo plazo. La estenosis de la anastomosis portal puede requerir angioplastia mediante portografía directa.
• Complicaciones biliares a largo plazo. Complicación frecuente del trasplante de donante vivo. La estenosis biliar afecta al 5-25% de los trasplantes pediátricos(17). Las manifestaciones habituales son disfunción del injerto con GGT elevada o colangitis recurrentes(18). Las estenosis pueden ser de la anastomosis biliodigestiva o estenosis más difusas secundarias a patología isquémica arterial; las primeras responden mejor a los procedimientos de dilatación con balón mediante colangiografía transparietohepática. Algunos de los pacientes con estenosis biliar no resuelta y riesgo de colangitis precisan antibioterapia oral prolongada.
• Posibilidad de obstrucción intestinal por bridas. Estos pacientes tienen riesgo de desarrollar obstrucción intestinal por bridas, cursa con vómitos. Puede requerir cirugía. Es importante asegurar adecuada hidratación iv.
Inmunosupresión. Fármacos empleados. Interacciones farmacológicas. Situaciones que pueden modificar los niveles. Profilaxis de insuficiencia suprarrenal
Los esquemas más habituales a largo plazo incluyen tacrolimus (inhibidor de calcineurina) con o sin esteroide.
Las pautas no son iguales en los diversos centros. En caso de aparición de efectos adversos relacionados con tacrolimus, como alergia alimentaria grave o hipertrofia ventricular, se sustituye tacrolimus por ciclosporina. El nivel de inmunosupresión deseado es mayor en los primeros meses postrasplante; a largo plazo, el objetivo es mantener al paciente con la mínima inmunosupresión necesaria para mantener función hepática normal, reduciendo los efectos secundarios. Los inhibidores de calcineurina se asocian con desarrollo de insuficiencia renal a largo plazo. En los pacientes con mayor riesgo de insuficiencia renal, se puede asociar otro inmunosupresor (mofetilmicofenolato), con el fin de reducir el nivel de tacrolimus. El sirolimus es otro inmunosupresor usado solo en pacientes seleccionados.
Presentaciones, forma de administración, monitorización, interacciones farmacológicas
Existen diferentes formas de presentación de tacrolimus; debe tomarse siempre respetando ayuno, 1 hora antes y 1 hora después. En niños pequeños, se utiliza la presentación en granulado para suspensión oral (Modigraf? sobres), cada 12 horas. En pacientes que son capaces de deglutir cápsulas, la presentación de liberación prolongada (Advagraf?) permite la administración en dosis única por la mañana, en ayunas, facilitando el cumplimiento terapéutico, especialmente en adolescentes. La dosificación se ajusta en función del nivel valle (extraído antes de la dosis) deseado, este objetivo de nivel cambia dependiendo del tiempo postrasplante y otros factores.
El ajuste de los inmunosupresores debe realizarse solo por los médicos especialistas encargados del manejo del paciente.
La ciclosporina (Sandimmun Neoral) se presenta en cápsulas y en solución pediátrica, se administra cada 12 horas. No precisa ayuno, se toma con comida. La dosificación se hace en función del nivel pico deseado, extraído a las 2 horas de la administración.
El corticoide se administra en dosis matutina única diaria o en días alternos (dependiendo del momento del trasplante).
Interacciones farmacológicas que modifican los niveles
Los inhibidores de calcineurina (tacrolimus y ciclosporina) son metabolizados por el citocromo P-450 en el hígado(19,20).
Algunos fármacos de uso frecuente, como macrólidos (azitromicina, eritromicina y claritromicina) o antifúngicos (fluconazol, itraconazol, ketoconazol) inhiben el citocromo P450, generando un aumento importante de los niveles de tacrolimus y ciclosporina, que pueden alcanzar rango tóxico.
Por el contrario, los fármacos que inducen el citocromo P450, como: fenobarbital, fenitoína, carbamacepina, isoniacida o rifampicina, reducen los niveles de anticalcineurínicos, pudiendo favorecer rechazo del injerto. En la medida de lo posible, se recomienda evitar dichos fármacos. En el caso de que sea imprescindible su uso, se contactará con el Servicio de Trasplante Hepático para ajustar la dosis de inmunosupresión.
Otras situaciones frecuentes que pueden modificar los niveles
La diarrea provoca un incremento de la absorción intestinal de tacrolimus, con aumento de su nivel, lo que unido a la posible deshidratación, incrementa el riesgo de insuficiencia renal aguda. Es importante contactar con el Servicio de Trasplante Hepático para ajustar la dosis.
Los vómitos pueden provocar descenso del nivel de inmunosupresores.
Profilaxis de insuficiencia suprarrenal
Los pacientes que reciben corticoterapia mantenida tienen riesgo de insuficiencia suprarrenal en situaciones de estrés, como cirugías e infecciones graves. Es aconsejable aumentar la dosis de corticoide (triplicar la producción fisiológica de cortisol) y volver a la dosis previa en cuanto se solvente la situación de riesgo.
Rechazo agudo y crónico(18,20)
El rechazo puede ser mediado por respuesta inmunológica celular o humoral. El rechazo agudo celular generalmente ocurre en los primeros días postrasplante, pero puede suceder en cualquier momento de la evolución, sobre todo, si hay incumplimiento terapéutico. Se manifiesta con disfunción hepática con GGT elevada. La confirmación es histológica. Responde bien al tratamiento con bolos de esteroides y aumento de inmunosupresión. El rechazo crónico ocurre en 5-10%, se caracteriza por: disfunción, la histología final es fibrosis, ductopenia y arteriopatía obliterativa. El manejo consiste en aumento o modificación de la inmunosupresión, asegurando el cumplimiento terapéutico. Es causa de pérdida del injerto y retrasplante. En los últimos años, se ha reconocido el rechazo mediado por anticuerpos(20) como causa de disfunción en el trasplante hepático.
Alteración renal
Los inhibidores de la calcineurina causan nefrotoxicidad, con disminución del filtrado glomerular y tubulopatía.
Existen otros factores implicados en el desarrollo de insuficiencia renal, como la patología renal preexistente (en: síndrome de Alagille, tirosinemia, nefrotoxicidad por quimioterapia en hepatoblastoma). Algunos pacientes reciben como tratamiento habitual, suplementos de bicarbonato o magnesio, y antihipertensivos. En cada revisión hospitalaria, se monitoriza la función renal y la tensión arterial.
Manejo práctico en Atención Primaria y Urgencias del problema renal
En los pacientes trasplantados hepáticos, es necesario evitar situaciones que pueden desencadenar insuficiencia renal aguda o agravar la disfunción renal preexistente.
Es importante garantizar una correcta hidratación y recordar que en estos niños, la exploración clínica no siempre se correlaciona con el grado de deshidratación; por ello, cuando presenten vómitos o diarrea, el manejo será más agresivo, con control de iones y gasometría e instauración de fluidoterapia iv, si no está garantizado un adecuado aporte oral.
En cuanto a los fármacos, los antiinflamatorios no esteroideos están contraindicados. Si es necesario el empleo de fármacos nefrotóxicos, se realizará con monitorización de niveles.
Infecciones(17)
Las infecciones tempranas, en el primer mes, están relacionadas con la cirugía y son de predominio bacteriano y fúngico. En el período comprendido entre el 1º y 6º mes postrasplante, debido al mayor grado de inmunosupresión requerido, son más frecuentes las infecciones por oportunistas como: Pneumocystis jirovecii, CMV, Epstein-Barr y por gérmenes adquiridos en la comunidad, que se pueden comportar con mayor agresividad que en niños sanos (rotavirus, virus sincitial respiratorio, influenza…). Los pacientes reciben profilaxis de infección por Pneumocystis durante 6-12 meses postrasplante con trimetroprín-sulfametoxazol. En caso de infección por virus Influenza, está indicado tratamiento con oseltamivir. Hay que recordar que el riesgo de gérmenes oportunistas reaparece si se requiere incremento importante de la inmunosupresión por rechazo. A partir del 6º mes, en el que desciende la inmunosupresión, se considera que el riesgo infeccioso, el tipo de infecciones y el manejo es similar al de los niños inmunocompetentes.
En pacientes con estenosis biliar que presentan clínica infecciosa sin foco, sobre todo, si se acompaña de disfunción del injerto, hay que pensar en la posibilidad de colangitis como causa del proceso.
Infecciones virales: CMV, Epstein-Barr (VEB) y varicela
Tanto el CMV como el VEB causan morbimortalidad en los pacientes con trasplante hepático y la infección puede ser primaria o por reactivación.
La infección por CMV(17) presenta clínica variable, desde: infección asintomática a cuadro viral inespecífico, neumonía, síntomas gastrointestinales o hepatitis. Ha sido asociado también con: rechazo, infección fúngica y pérdida del injerto. El VEB tiene una gran importancia en el trasplante pediátrico, la infección puede cursar asintomática, como mononucleosis o desencadenar un síndrome linfoproliferativo (PTLD). Los pacientes reciben profilaxis inicial de infección por CMV y VEB con ganciclovir iv y, posteriormente, con valganciclovir oral (3-6 meses).
La varicela se trata con aciclovir oral. En caso de varicela complicada, es aconsejable el ingreso hospitalario.
Normas generales para los pacientes: correcta higiene de manos, evitar ingesta de frutas no peladas o verduras sin cocinar, y carne poco cocinada.
Síndrome linfoproliferativo (PTLD) y otros tumores
PTLD es una de las complicaciones más graves del trasplante hepático, la incidencia (6%) ha disminuido en los últimos años. PTLD precoz se relaciona con infección VEB, los factores de riesgo son la seronegatividad pretrasplante y el grado elevado de inmunosupresión.
La actual clasificación WHO 2017, divide PTLD en cuatro categorías: no destructivo, polimorfo, monomorfo y linfoma. La clínica es muy variada, se necesita un alto grado de sospecha.
Los datos de alarma son: cuadros de mononucleosis infecciosa, adenopatías, hipertrofia amigdalar con: clínica de obstrucción (ronquido), diarrea o fiebre prolongada, neumonitis, anorexia, pérdida de peso y hepatoesplenomegalia. La monitorización seriada de la carga viral del VEB permite disminuir la inmunosupresión cuando se observan cargas virales elevadas, dato que, en ocasiones, precede al desarrollo de PTLD. El tratamiento inicial de PTLD es la disminución o retirada de tacrolimus/ciclosporina, rituximab en casos más avanzados y quimioterapia en caso de linfomas.
Los trasplantados hepáticos tienen riesgo aumentado de otros tumores. Es importante la protección solar, por el riesgo aumentado de melanoma y otros cánceres en la piel.
Vacunación(21,22)
Postrasplante no pueden recibir vacunas de gérmenes vivos atenuados; por tanto, no pueden recibir las vacunas: triple vírica, varicela, rotavirus, BCG, fiebre amarilla, fiebre tifoidea oral, polio oral y la vacuna influenza atenuada intranasal.
Se continuará con la inmunización comenzada pretrasplante a partir del 4º-6º mes postrasplante (incluso desde el 2º mes)(20). Anualmente en otoño, deben recibir la vacuna antigripal inactivada; se puede administrar a partir del mes postrasplante si coincide con epidemia de gripe. Es necesario asegurar protección frente al virus de hepatitis A y B, con revacunación si fuera necesario. Están indicadas las vacunas frente a: neumococo 13 v y 23 v, meningococo B, meningococo A, C, W, Y; así como, las vacunas frente a papiloma humano (también en varones). Pueden requerir dosis de refuerzo.
En los últimos años, debido a los brotes de sarampión y a las menores tasas de vacunación en algunos países, existe una preocupación creciente por la posibilidad de infección postrasplante(22). Los expertos están tratando de elaborar recomendaciones sobre la vacunación en función del grado de inmunosupresión.
Vacunación de los convivientes. Deben recibir la vacuna antigripal inactivada todos los años, se aconseja que los contactos no inmunes reciban la vacuna triple vírica y varicela. Si aparece exantema tras la vacunación frente a varicela, se debe evitar el contacto con el trasplantado hasta que desaparezca. Están contraindicadas en convivientes, la vacuna de la polio oral y la antitifoidea oral. Los hermanos pueden recibir vacunación frente a Rotavirus, pero extremando las medidas de higiene.
Alergia alimentaria de novo
Afecta aproximadamente al 15%, preferentemente niños menores de 2 años que reciben tacrolimus. Mejoran con el cambio a ciclosporina. Un pequeño porcentaje de trasplantados hepáticos desarrollan esofagitis eosinofílica. Es frecuente también la queilitis.
Crecimiento(10), retraso puberal, obesidad
Después del trasplante, se produce “catch-up” de crecimiento, pero un 20% de los niños trasplantados tienen una talla final inferior al percentil 5. Los factores relacionados con la alteración del crecimiento son: peor estado nutricional pretrasplante, retraso de talla al trasplante, retrasplante, uso prolongado de esteroides y patología de base diferente a la atresia. Es frecuente el retraso puberal. La obesidad junto con la hiperlipidemia y la hipertensión, son factores de riesgo cardiovascular. Es necesario promover hábitos de vida saludables (dieta apropiada, ejercicio regular) en esta población de riesgo.
Adolescentes
Durante la adolescencia existe un alto riesgo de incumplimiento terapéutico que puede comprometer el injerto hepático.
En las revisiones, es necesario identificar factores de riesgo psicosociales y familiares. La simplificación del tratamiento (mínimo número de fármacos, inmunosupresores de liberación prolongada) y el uso de alarmas pueden facilitar el cumplimiento. Es conveniente incrementar la frecuencia de las revisiones en los casos de falta de adherencia y fomentar la colaboración familiar y el abordaje multidisciplinar. En esta etapa de la vida, se debe insistir en evitar ingesta de alcohol y empleo de drogas. Es importante alertar sobre el riesgo de enfermedades de transmisión sexual y de embarazo.
Las jóvenes que reciben mofetilmicofenolato deben ser informadas sobre la necesidad de emplear métodos anticonceptivos seguros por el riesgo de teratogenicidad.
El proceso de transición al Servicio de adultos requiere una estrecha colaboración entre los equipos infantil y de adultos.
Calidad de vida
Existen muchos factores implicados en la calidad de vida y los análisis, en este sentido, no son sencillos. En general, la calidad de vida postrasplante es similar a la de otras enfermedades crónicas e inferior a la de la población general. Es necesario identificar los factores que se pueden modificar para mejorar la calidad de vida.
Problemas dentales(20)
Son frecuentes: caries, gingivitis y malposición dental. La ciclosporina causa hipertrofia gingival. Se recomiendan controles periódicos por odontólogos y profilaxis antibiótica en caso de manipulaciones invasivas.
Cometido del pediatra de Atención Primaria
en el seguimiento de niños con hepatopatía crónica
y trasplante hepático
El cometido del pediatra en Atención Primaria es la identificación de recién nacidos con colestasis. En niños con hepatopatía colestática, la función principal es: tratamiento antibiótico precoz de las infecciones, asegurar una correcta vacunación, evitar medicamentos que causen riesgo de hemorragia, apoyar la correcta alimentación y medicación de sostén. En el postrasplante, el papel del pediatra de Atención Primaria será, desde el punto de vista preventivo: asegurar la correcta vacunación del paciente y sus convivientes, reforzar las recomendaciones generales encaminadas a promover hábitos saludables (dieta adecuada, ejercicio regular, protección solar, educación sexual) e insistir en el cumplimiento terapéutico en adolescentes. Desde el punto de vista del tratamiento, su cometido será: la identificación de problemas que puedan requerir atención hospitalaria y el manejo de las infecciones habituales que no precisen tratamiento intravenoso. Es importante conocer las interacciones más frecuentes de la medicación inmunosupresora.
Bibliografía
Los asteriscos muestran el interés del artículo a juicio del autor.
1. Fawaz R, Baumann U, Ekong U, Fischler B, Hadzic N, Mack CL, et al. Guideline for the evaluation of cholestatic jaundice in infants: Joint Recommendations of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition and the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition. J Pediatr Gastroenterol Nutr. 2017; 64: 154-68.
2.** Shneider BL, Moore J, Kerkar N, Magee JC, Ye W, Karpen SJ, et al. Childhood Liver Disease Research Network. Initial assessment of the infant with neonatal cholestasis-Is this biliary atresia? PLoS One. 2017; 12: e0176275.
3. Fanna M, Masson G, Capito C, Girard M, Guerin F, Hermeziu B, et al. Management of biliary atresia in France 1986 to 2015: long-term results. J Pediatr Gastroenterol Nutr. 2019; 69: 416-24.
4.** Shneider BL, Magee JC, Karpen SJ, Rand EB, Narkewicz MR, Bass LM, et al. Childhood Liver Disease Research Network (ChiLDReN). Total Serum Bilirubin within 3 Months of Hepatoportoenterostomy Predicts Short-Term Outcomes in Biliary Atresia. J Pediatr. 2016; 170: 211-7.e1-2.
5. Tessier MEM, Shneider BL, Brandt ML, Cerminara DN, Harpavat S. A phase 2 trial of N-Acetylcysteine in Biliary atresia after Kasai portoenterostomy. Contemp Clin Trials Commun. 2019; 15: 100370.
6. Duché M, Ducot B, Ackermann O, Guérin F, Jacquemin E, Bernard O. Portal hypertension in children: High-risk varices, primary prophylaxis and consequences of bleeding. J Hepatol. 2017; 66: 320-7.
7. Turnpenny PD, Ellard S. Alagille syndrome: pathogenesis, diagnosis and management. Eur J Hum Genet. 2012; 20: 251-7.
8. Davit-Spraul A, Gonzales E, Baussan C, Jacquemin E. Progressive familial intrahepatic cholestasis. Orphanet J Rare Dis 2009; 4: 1-1172.
9. Thébaut A, Nemeth A, Le Mouhaër J, Scheenstra R, Baumann U, Koot B, et al. Oral Tocofersolan Corrects or Prevents Vitamin E Deficiency in Children With Chronic Cholestasis. J Pediatr Gastroenterol Nutr. 2016; 63: 610-5.
10. Thébaut A, Debray D, Gonzales E. Physiopathology and management of cholestatic pruritus in children. Arch Pediatr. 2017; 24: 682-8.
11. Shneider BL, Spino C, Kamath BM, Magee JC, Bass LM, Setchell KD, et al. Childhood Liver Disease Research Network. Placebo-Controlled Randomized Trial of an Intestinal Bile Salt Transport Inhibitor for Pruritus in Alagille Syndrome. Hepatol Commun. 2018; 2: 1184-98.
12.** Mieli-Vergani G, Vergani D, Czaja AJ, Manns MP, Krawitt EL, Vierling JM, et al. Autoimmune hepatitis. Nat Rev Dis Primers. 2018; 4: 18017.
13. Efe C, Taii HA, Ytting H, Aehling N, Bhanji RA, Hagström H, et al. Tacrolimus and Mycophenolate Mofetil as Second-Line Therapies for Pediatric Patients with Autoimmune Hepatitis. Dig Dis Sci. 2018; 63: 1348-54.
14.** Socha P, Janczyk W, Dhawan A, Baumann U, D’Antiga L, Tanner S, et al. Wilson’s Disease in Children: A Position Paper by the Hepatology Committee of the European Society for Paediatric Gastroenterology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutr. 2018; 66: 334-44.
15.** Ng V, Alonso EM, Bucuvalas JC, Cohen G, Limbers CA, Varni JW, et al. For the Studies of Pediatric Liver Transplantation (SPLIT) Research Group. Health status of children alive 10 years after pediatric liver transplantation performed in the US and Canada: Report of the studies of pediatric liver transplantation experience. J Pediatr. 2012; 160: 820-6.
16.** Andrés AM, López Santamaría M, Hernández F. Técnicas quirúrgicas. En: Paloma Jara, ed. Tile Von Spain S.L; 2013. p. 799-819.
17.** Kelly DA, Bucuvalas JC, Alonso EM, Karpen SJ, Allen U, Green M, et al. Long-Term medical management of the pediatric patient after liver transplantation: 2013 Practice guideline by the American Association for the study of liver diseases and the American Society of Transplantation. Liver Transpl. 2013; 19: 798-825.
18.** Kerkar N, Lakhole A. Pediatric liver transplantation: a North American perspective. Expert Rev Gastroenterol Hepatol. 2016; 10: 949-59.
19. McGuire BM, Rosenthal P, Brown CC, Busch AMH, Calcatera SM, Claria RS, et al. Long-term management of the liver transplant patient: Recommendations for the Primary Care Doctor. Am J Transplant. 2009; 9: 1988-2003.
20.** Hassan S, Ng VL, Aqul A. It takes a village: primary care of the pediatric liver transplant recipient. Curr Opin Pediatr. 2019; 31: 636-44.
21. Vacunación en niños candidatos y receptores de trasplante hepático. En: Manual de Trasplante Hepático. Paloma Jara. Tile Von Spain S.L.; 2016. p. 123-35.
22. Suresh S, Upton J, Green M, Pham-Huy, Posfay-Barbe KM, Michaels MG, et al. Live vaccines after pediatric solid organ transplant: Proceedings of a consensus meeting, 2018. Pediatr Transplant; 2019. p. 23.
Bibliografía recomendada
- Sundaram SS, Mack CL, Feldman AG, Sokol RJ. Biliary atresia: Indications and timing of liver transplantation and optimization of pretransplant care. Liver Transpl. 2017; 23: 96-109.
Artículo de revisión, describe el proceso de cuidados e indicación de trasplante del niño con atresia biliar.
- Fanna M, Masson G, Capito C, Girard M, Guerin F, Hermeziu B, et al. Management of biliary atresia in France 1986 to 2015: long-term results. J Pediatr Gastroenterol Nutr. 2019; 69: 416-24.
Experiencia en Francia con 1.428 pacientes con atresia biliar. El 94% recibieron Kasai, el 6% no operados por detección demasiado tardía. La edad media de operación fue de 59 días, no hubo mejoría en la edad de detección a lo largo de 3 décadas. Un 39% de niños evolucionaron con bilirrubina normal después del Kasai. La supervivencia con hígado propio fue de 41% a los 5 años, 35% a los 10 años y 26% a los 20 años. En los operados en el primer mes de vida, la supervivencia con hígado propio fue de 38% a los 25 años.
La supervivencia de niños con atresia que recibieron trasplante hepático fue de 79% a los 28 años. En el periodo reciente, la supervivencia del trasplante fue mayor (87% a los 5 años). Con Kasai y trasplante, si es necesario, el pronóstico de supervivencia de niños con atresia biliar es de 87%.
- Bull LN, Thompson RJ. Progressive Familial Intrahepatic Cholestasis. Clin Liver Dis. 2018; 22: 657-69.
Artículo de revisión actualizado de los diferentes tipos de cholestasis intrahepática familiar por defectos de: BSEP, FIC1, MDR3, TJP2, FXR y MYO5B.
- Kelly DA, Bucuvalas JC, Alonso EM, Karpen SJ, Allen U, Green M, et al. Long-Term medical management of the pediatric patient after liver transplantation: 2013 Practice guideline by the American Association for the study of liver diseases and the American Society of Transplantation. Liver Transpl. 2013; 19: 798-825.
Guía práctica extensa de recomendaciones clínicas de la Asociación Americana. Expone los problemas principales del trasplante hepático pediátrico, y las recomendaciones de seguimiento y manejo en cada uno de ellos.
- Hassan S, Ng VL, Aqul A. It takes a village: primary care of the pediatric liver transplant recipient. Curr Opin Pediatr. 2019; 31: 636-44.
Artículo que expone, de forma concisa, aspectos de la inmunosupresión y complicaciones postrasplante.
- Kerkar N, Lakhole A. Pediatric liver transplantation: a North American perspective. Expert Rev Gastroenterol Hepatol. 2016; 10: 949-59.
Artículo en el que se abordan las indicaciones del trasplante y las complicaciones después del trasplante hepático, aportando una idea clara global.
| Caso clínico |
|
Primera consulta postrasplante en Atención Primaria de una paciente de 14 meses trasplantada hace 6 meses. La madre tiene varias dudas en relación con las vacunas de la paciente y su entorno. En la casa conviven la paciente, dos hermanos y los padres, todos ellos sanos. Proceden de Ecuador, pero los hijos han nacido aquí y han recibido el esquema vacunal español.
|
