 |
| Temas de FC |
E. Baselga Torres*, C.E. Alarcón Pérez**
*Dermatóloga Pediatra. Jefe del Servicio de Dermatología Pediátrica. **Dermatólogo. Máster en Dermatología Pediátrica. Hospital Sant Joan de Déu Barcelona
| Resumen
Las anomalías vasculares son un motivo de consulta muy frecuente en Pediatría y en Dermatología, que dada la diversa nomenclatura que se ha usado para intentar clasificarlas, todavía generan muchas dudas diagnósticas. Diferenciar entre una lesión tumoral y otra malformativa es importante, puesto que el abordaje diagnóstico, terapéutico y pronóstico va a ser muy diferente. Las malformaciones vasculares constituyen anomalías del desarrollo de vasos de distinto tipo que están presentes desde el nacimiento y pueden permanecer toda la vida. Por otro lado, los tumores son lesiones proliferativas que no siempre son congénitos, pueden involucionar por sí solos y dejar secuelas estéticas y funcionales. Ambos grupos se pueden asociar a diversos trastornos con elevada morbi-mortalidad, por lo que un adecuado reconocimiento impactará en la vida del afectado. Los médicos de Atención Primaria y pediatras son los facultativos de primera línea que pueden detectar, desde una simple marca de nacimiento que no requiere exploraciones complementarias hasta identificar los casos graves que necesitan intervenciones prioritarias. |
| Abstract
Vascular anomalies represent a highly frequent reason for consultation in pediatric and dermatology clinics. There are still many diagnostic queries among hospital and primary care physicians due to previously used wide and di-verse nomenclatures aiming to classify them into a homogeneous group. Differentiating between a tumor and malformation is important since the diagnostic and therapeutic approach will differ, as well as the prognosis. Vascular malformations result from abnormal morphogenesis of different types of vessels, they are present at birth and may remain lifelong. On the other hand, tumors are proliferative lesions that are not always congenital and may involute rapidly, leaving aesthetic and functional sequelae in certain cases. Both may be associated to various diseases of high morbi-mortality, where prompt recognition is essential. Primary care physicians and pediatricians represent a first-line detection point for differentiating between a single birthmark with no demand of strict follow-up, to recognizing severe cases where priority intervention is required. |
Palabras clave: Anomalías vasculares; Hemangioma; Propranolol; Mancha en vino de Oporto; Sobrecrecimiento.
Key words: Vascular anomalies; Hemangioma; Propranolol; Port wine stain; Overgrowth.
Pediatr Integral 2021; XXV (3): 128.e1 – 128.e22
Anomalías vasculares
Introducción
Las anomalías vasculares constituyen un espectro de entidades que se manifiestan desde temprana edad y van, desde una simple marca de nacimiento hasta diversos trastornos con elevada morbi-mortalidad. La nomenclatura empleada para denominar estas enfermedades ha sido por muchos años confusa y, por ello, la Sociedad Internacional para el estudio de anomalías vasculares (ISSVA) propone separar las anomalías vasculares en dos grandes grupos: los tumores y las malformaciones vasculares (Tabla I)(1). Los avances en la genética de estas anomalías han permitido dilucidar las diferentes vías moleculares alteradas y posibles dianas terapéuticas.

En este artículo, se revisarán los tumores y malformaciones vasculares más frecuentes en la infancia.
Tumores vasculares benignos
Hemangioma infantil
Los hemangiomas infantiles suelen verse como una mancha rosada al momento de nacer, proliferan a las pocas semanas de vida y se estabilizan alrededor de los 4 a 6 meses; la necesidad de tratamiento va a depender de la localización y de las posibles complicaciones asociadas.
El hemangioma infantil (HI) es el tumor más frecuente en la infancia, afectando aproximadamente del 4 al 10% de lactantes(2). Son más frecuentes en: sexo femenino y raza caucásica, niños prematuros, recién nacidos con bajo peso, edad materna avanzada, gestación múltiple, placenta previa, y preeclampsia. La mayoría se localizan en la piel y mucosas. Las localizaciones extracutáneas más frecuentes son el hígado y la vía aérea(3).
Curso clínico. Los hemangiomas tienen una historia natural característica de crecimiento rápido e involución, que permite sospechar el diagnóstico.
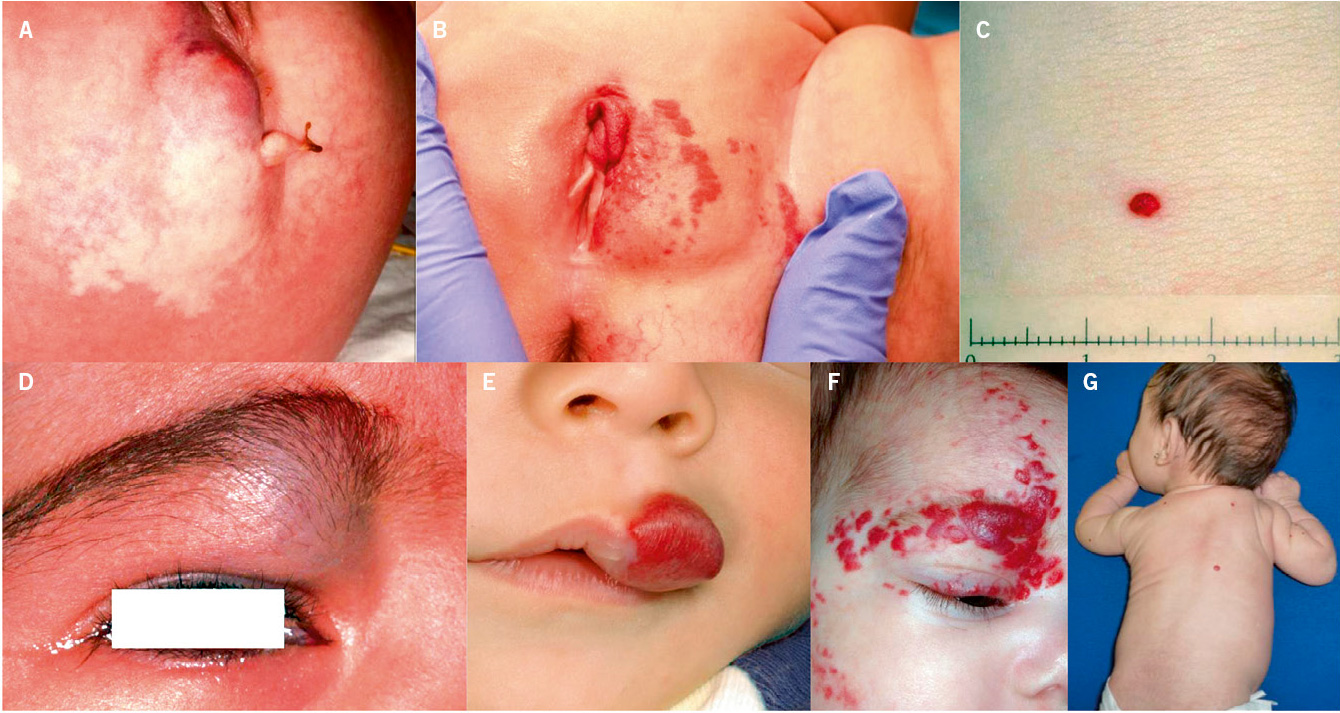
Figura 1. Clasificación de los hemangiomas infantiles según la fase de crecimiento, profundidad de los vasos, forma y distribución.
A. Precursor de hemangioma: lesión precursora de un hemangioma infantil en región lumbar representada por un área de vasoconstricción; B. Hemangioma infantil abortivo o de proliferación mínima: se suelen apreciar como una escasa proliferación de pápulas o placas rojizas con telangiectasias en su superficie; C. Hemangioma focal superficial: clínicamente se ven como pápulas o placas de forma redondeada (focal significa que se puede trazar con un compás), de color rojo brillante con superficie lobulada o lisa; D. Hemangioma focal profundo: se exponen como tumoraciones azuladas o del color de la piel normal, redondeadas, algunas veces con telangiectasias en su superficie; E. Hemangioma focal mixto: nódulo focal que tiene un componente superficial rojo brillante y un componente profundo que aporta volumen; F. Hemangioma segmentario superficial: este hemangioma tiene solo componente superficial rojo, ocupa una superficie geográfica de bordes imprecisos y no podría ser trazado con un compás como los hemangiomas focales; G. Hemangioma multifocal: los HI pueden ser múltiples, en estos casos, son generalmente focales, por lo que se denominan multifocales.
Al nacimiento, no suelen estar presentes o como mucho hay una lesión precursora en forma de mácula rosada / telangiectásica o un área de vasoconstricción (Fig. 1A). Durante las primeras semanas de vida, inician una fase proliferativa rápida hasta los 3-4 meses de edad, seguida de un periodo de estabilidad y, a partir del año de edad, una fase involutiva que se prolonga hasta los 4-5 años. La involución no es sinónimo de desaparición, puesto que puede quedar alguna secuela en forma de telangiectasias, piel redundante o piel anetodérmica(4). Ante un hemangioma concreto es difícil predecir la duración de cada fase. Si bien, los HI profundos y segmentarios suelen tener una fase proliferativa más prolongada, hay hemangiomas llamados abortivos o de proliferación mínima, que tienen escasa o nula proliferación (Fig. 1B)(5). El crecimiento tardío, más allá del año de edad, es inusual aunque está descrito(6).
Manifestaciones clínicas. El aspecto de los hemangiomas depende de su profundidad (superficiales, mixtos y profundos) y de su patrón de distribución (focales o segmentarios) (Figs. 1C-F)(2). Los HI pueden ser múltiples y, en estos casos, son generalmente focales, por lo que se denominan multifocales (Fig. 1G).
Asociaciones. Los hemangiomas segmentarios de la cabeza y cuello, y de la región lumbosacra, pueden asociar alteraciones estructurales subyacentes que se denominan con los acrónimos PHACES y LUMBAR/PELVIS/SACRAL, respectivamente (Figs. 2A y B y tabla II).
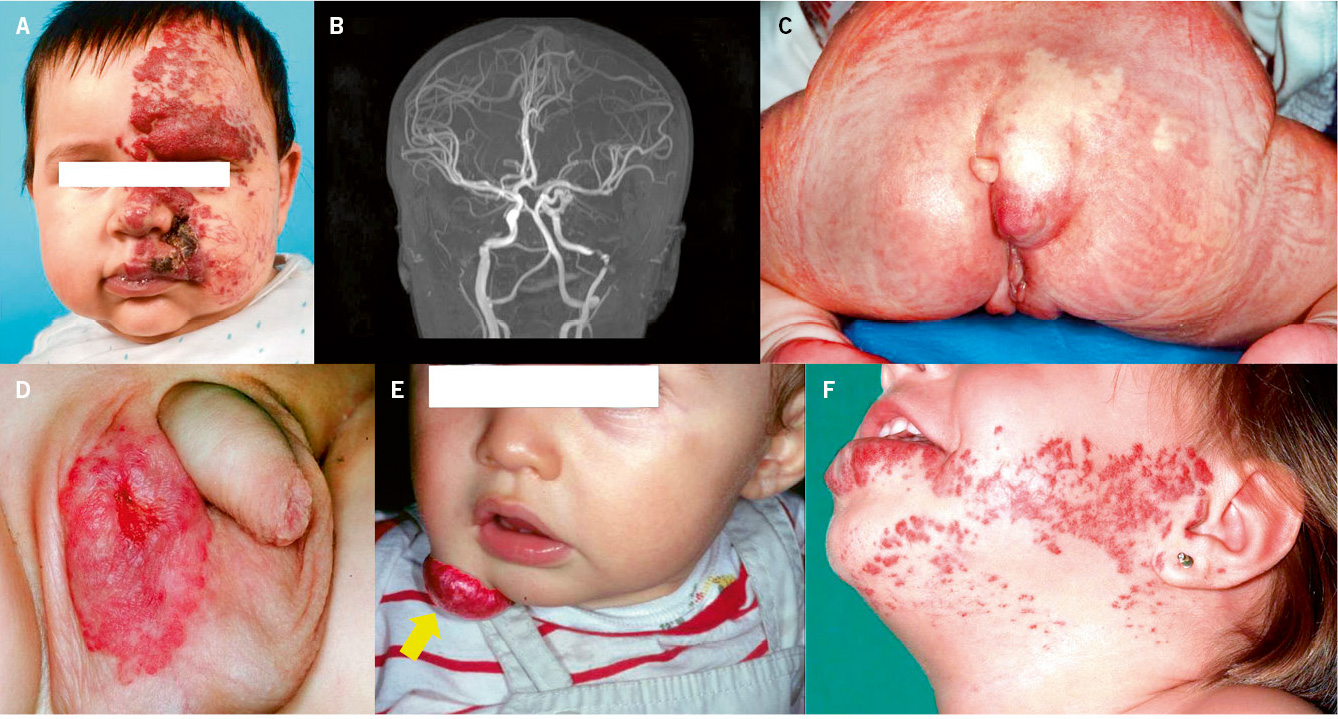
Figura 2. Asociaciones y complicaciones de los hemangiomas infantiles. A. Hemangioma infantil segmentario y, en este caso, ulcerado de más de 5 cm en la cara. Esta paciente tiene riesgo de síndrome PHACES; B: corte tridimensional de la RM cerebral de la paciente de la figura 2A, en el que se reportaba estenosis de la arteria carótida izquierda en el segmento cervical y una configuración alterada del polígono de Willis. También, tenía otras alteraciones en fosa posterior en relación con síndrome de PHACES; C. Síndrome LUMBAR/PELVIS/SACRAL: hemangioma infantil mixto de más de 5 cm con defectos de la línea media, localizado en región lumbar sobre la línea media; D. Hemangioma ulcerado: se suelen ulcerar en la porción central. Con frecuencia, se ulceran los que se localizan en áreas de roce y trauma, como: región anogenital, pliegues, labios, frente y nariz; E y F. Hemangioma infantil del área de la barba: pueden ser focales mixtos (E) o segmentarios (F). Se puede asociar a hemangiomas subglóticos con compromiso de la vía área, especialmente los que se localizan en labio y encía inferior, mentón y cara anterior del cuello. Clínicamente, el paciente puede presentar tos, estridor inspiratorio, ronquera o apneas obstructivas mientras duerme.

Las alteraciones asociadas y exploraciones complementarias recomendadas en estos casos, aparecen en la tabla II(7,8). Los hemangiomas segmentarios de la zona de la barba, además, pueden asociar hemangiomas subglóticos con compromiso de la vía área, especialmente los localizados en: labio inferior, encía inferior, mentón y cara anterior del cuello (Figs. 2E y F).
Los niños con hemangiomas multifocales tienen mayor riesgo de tener hemangiomas hepáticos. Por ello, se recomienda realizar una ecografía hepática a los niños con 5 o más hemangiomas focales.
Diagnóstico. El diagnóstico es clínico, por medio de la exploración física y la historia evolutiva. Los hemangiomas profundos son los que pueden ofrecer más dudas. El estudio anatomopatológico y, sobre todo, el inmunohistoquímico permiten diferenciar el HI de otros tumores vasculares, ya que es el único tumor vascular que expresa el marcador GLUT-1, una proteína transportadora de glucosa(2).
Diagnóstico diferencial. La lesión precursora de los HI y los hemangiomas “abortivos” se puede confundir con malformaciones capilares, nevus anémicos e, inclusive, traumatismos. Los HI profundos pueden simular un quiste dermoide, un glioma, un lipoma o un pilomatrixoma, entre otros. El eco-Doppler puede ser útil en estos casos, para identificar la naturaleza vascular de la lesión. En los casos multifocales, deben diferenciarse de la linfangioendoteliomatosis multifocal, el síndrome del nevus azul en tetina de goma (síndrome de Bean), los granulomas piogénicos múltiples congénitos y la histiocitosis de células de Langerhans(2,5).
Tratamiento. La mayoría de los hemangiomas no requieren tratamiento, porque involucionan espontáneamente. Sin embargo, entre un 12-20% de los HI van a requerir tratamiento por compromiso estético o funcional. Así, las indicaciones para tratar un HI son:
• HI potencialmente mortal o que ponga en peligro alguna capacidad funcional (p. ej.: hemangioma de la vía área, perioculares, hemangiomas de gran tamaño, hemangiomas que impiden la succión, etc.).
• HI ulcerado (Fig. 2D) con dolor y/o ausencia de respuesta a las medidas básicas del cuidado de heridas.
• HI con riesgo de desfiguración o cicatrices permanentes. Esta es la indicación de tratamiento más frecuente y difícil de tomar, porque obliga a predecir la repercusión psicológica o el residuo que va a dejar un hemangioma en concreto. Por regla general, van a tener consideración estética: hemangiomas de gran tamaño en cualquier localización, hemangiomas que afectan la zona central de la cara y nariz, hemangiomas que distorsionan los labios o que afectan la areola mamaria. En este sentido, los hemangiomas segmentarios y los hemangiomas con un componente superficial grueso o de borde abrupto, suelen dejar más secuela(4,9).
Si bien, todos los HI involucionan, pueden dejar cambios en el aspecto de la piel o piel redundante(4,10,11).
En la actualidad, el propranolol por vía oral es el único tratamiento aprobado para esta indicación. El tratamiento debe iniciarse idealmente durante el periodo de crecimiento más rápido de los HI; es decir, entre el primer y el tercer mes de vida. Si bien, su uso está aprobado a partir de las 5 semanas de vida, los estudios en menores a 5 semanas indican que también es seguro en este grupo de edad e incluso en los prematuros que lo requieran. Antes de iniciar el tratamiento, se debe realizar una exploración física completa con auscultación cardiopulmonar para descartar alteraciones del ritmo cardíaco o broncoespasmo. Se inicia ambulatoriamente a 1 mg/kg dividido en dos dosis, con control de tensión arterial y frecuencia cardiaca a la hora, y a las dos horas, para aumentar semanalmente, realizando la misma monitorización hasta la dosis aprobada de 3 mg/kg/día dividida en dos dosis. La respuesta al propranolol es rápida y prácticamente universal; de manera que, a veces, se utiliza como prueba diagnóstica. El 60% de los hemangiomas se resuelven completamente con 6 meses de tratamiento. Si la resolución no es completa, puede ser útil alargar el tratamiento hasta el año de edad. Al suspender el tratamiento, es posible observar una leve recoloración del hemangioma o incluso un recrecimiento, que obligue a reiniciar el medicamento.
Recientemente, el grupo investigador de hemangiomas (HIG) ha publicado unas recomendaciones de manejo de los hemangiomas infantiles durante la pandemia del COVID que es extrapolable a situaciones en las cuales no se pueda valorar de manera presencial al paciente. En estos casos, en ciertos pacientes de bajo riesgo, se puede considerar el inicio de betabloqueadores sin control presencial de constantes, siempre y cuando tengamos una exploración cardiorrespiratoria normal. En estos casos, se recomienda iniciar a dosis más bajas (0,5 mg/kg/día dividida en dos dosis) para ir incrementando, cada 3 a 4 días, de a 0,5 mg/kg/día hasta la dosis final aprobada. El principal efecto adverso del propranolol es la hipoglucemia que puede suceder en lactantes pequeños y con ayunos prolongados. Por ello, es importante insistir a los padres que se administre coincidiendo con una comida; que no pasen más de 6 horas sin ninguna toma y, en caso de rechazo a la ingesta, suspender el propanolol. Si hay contraindicación para el uso de betabloqueadores o reacciones adversas en HI de alto riesgo, están indicados los corticoides sistémicos(10,11).
Otra alternativa terapéutica no aprobada para HI pequeños, superficiales o muy iniciales, pero que comportan algún riesgo estético por su localización es el timolol tópico. Se suele emplear colirio de timolol al 0,5%, sin sobrepasar dos gotas dos veces al día, porque a dosis superiores se ha demostrado absorción sistémica(12). No se recomienda usar en casos de HI profundos, porque es menos efectivo y hay mayor riesgo de absorción sistémica. La cirugía y la terapia láser son opciones, principalmente, para el manejo de las secuelas, no obstante el láser de colorante pulsado puede ser útil para el manejo de HI ulcerados en combinación con el propranolol(10).
Prevención. Para prevenir secuelas o detectar complicaciones asociadas a los hemangiomas, es muy importante estratificar a los hemangiomas según el riesgo de complicaciones y, en caso de requerir tratamiento, iniciarlo cuanto antes (Tabla III).

Hay HI que debemos seguir más de cerca y/o referir al especialista, y hay otros que prácticamente no requieren un seguimiento específico. En hemangiomas de riesgo, en fase proliferativa temprana, se recomienda seguimiento clínico cada 10-15 días, mientras que en hemangiomas estabilizados, el seguimiento puede ser trimestral(10).
En Pediatría primaria, es muy importante identificar los hemangiomas que requieren remisión inmediata al especialista y que exista una vía de comunicación rápida con el mismo, o con el centro con experiencia en HI. En este sentido, se ha publicado un hemangioma referral score IHReS (www.ihscoring.com/es), que es una herramienta validada desarrollada con dermatólogos y pediatras, con el objetivo de facilitar el reconocimiento de los HI que requieren derivación(13). Se tienen en cuenta: características y localización del propio HI, así como edad del paciente, ya que no es lo mismo un HI en un lactante de un mes en plena fase proliferativa, que un HI en un lactante de 6 meses en el que es improbable que crezca más.
Hemangiomas congénitos
Los hemangiomas congénitos a diferencia de los hemangiomas infantiles están plenamente desarrollados al nacer y no responden al tratamiento con propranolol.
Los hemangiomas congénitos (HC) están presentes y desarrollados al momento de nacer, y son negativos para el marcador de inmunohistoquímica GLUT-1. Dependiendo de la historia natural y de las características clínicas, se reconocen tres formas clínicas de HC: hemangiomas congénitos rápidamente involutivos (RICH), hemangiomas congénitos parcialmente involutivos (PICH) y hemangiomas congénitos no involutivos (NICH). A pesar de que esta clasificación es descriptiva y engloba a la gran mayoría de casos, el espectro es más amplio, pues recientemente se han descrito presentaciones atípicas: formas multifocales; NICH con crecimiento tardío después de años de estabilidad; y comportamientos inusuales en algunos PICH(14,15).
Epidemiología. Su incidencia es desconocida. No tiene predilección por sexo ni los factores de riesgo del HI.
Fisiopatología. Son el resultado de mutaciones activadoras en mosaico en GNAQ (Gln209) o GNA11 (Gln209)(16).
Manifestaciones clínicas. Se ilustran en las figuras 3A-E.

Figura 3. Manifestaciones clínicas de otros tumores vasculares diferentes al hemangioma infantil. A. Hemangiomas congénitos rápidamente involutivos (RICH): se presenta como una tumoración gríseo-violácea con telangiectasias gruesas en su superficie, rodeados por un borde pálido de vasoconstricción. La superficie es lisa y el centro puede estar ulcerado o atrófico. Los de gran tamaño suelen tener un vaso nutricio de gran tamaño. Se localizan de predominio en la cabeza y en el cuello o en las extremidades proximales a las articulaciones. Usualmente son únicos; B. RICH: involucionan rápidamente entre 6 a 18 meses de edad, dejando cambios mínimos; C. Hemangiomas congénitos no involutivos (NICH): se presentan en forma de placa aplanada con telangiectasias gruesas que crecen en proporción al niño y permanecen más o menos inmodificados toda la vida; D. Hemangiomas congénitos parcialmente involutivos (PICH): son lesiones que inicialmente son como RICH y que involucionan rápidamente, pero solo parcialmente, dejando una lesión similar a un NICH (figura 3, E); F. Angioma en penacho: placa de aspecto vascular similar al RICH, más sólida e infiltrada con hipertricosis y de superficie irregular con aspecto de “adoquín”. Puede haber hiperhidrosis localizada y ser dolorosas al tacto; G. Granuloma piógeno: suele manifestarse como una pápula rojiza de superficie lisa y brillante de crecimiento rápido, generalmente pediculadas, que sangran de manera prolongada tras el mínimo roce. En algunos casos, la base angosta se puede estrangular lo que genera necrosis y resolución de la lesión. Son frecuentes en cabeza y cuello; H. Granuloma piógeno: no es infrecuente que aparezcan sobre la superficie de las manchas en vino de Oporto*; I. Hemangioendotelioma kaposiforme: cuando afecta la piel, se presenta como una lesión única tipo placa eritematoviolácea indurada o como una masa sólida y profunda. Se localiza con más frecuencia en cuello, tronco y extremidades. *Groesser L, Peterhof E, Evert M, Landthaler M, Berneburg M, Hafner C. BRAF and RAS Mutations in Sporadic and Secondary Pyogenic Granuloma. J Invest Dermatol. 2016;136(2):481-6.
Diagnóstico. El diagnóstico es clínico, por medio de la exploración física y la historia evolutiva. El estudio anatomopatológico no diferencia entre RICH, NICH o PICH, pero tienen una histología diferente de los HI y son tumores GLUT-1 negativos. El Eco-Doppler muestra una lesión de flujo alto e inclusive puede ser detectado de manera prenatal.
Complicaciones. Los RICH grandes pueden causar insuficiencia cardiaca de alto flujo, también trombocitopenia transitoria que suele recuperarse en un plazo de 2-3 semanas y que no debe confundirse con el fenómeno de Kasabach-Merritt (FKM). En ocasiones, pueden ulcerarse centralmente y sangrar profusamente.
Tratamiento. No suelen requerir tratamiento. No responden ni a corticoides ni a propranolol. En los RICH de gran tamaño o con complicaciones, se han realizado embolizaciones y también tratamiento con rapamicina. Las telangiectasias residuales se pueden tratar con láser de colorante pulsado.
Angioma en penacho
El angioma en penacho es de curso benigno, pero puede causar trombocitopenia grave y coagulopatía de consumo (fenómeno de Kasabach-Merritt).
El angioma en penacho (AP) o angioblastoma de Nakagawa es un tumor vascular infrecuente que puede presentarse de manera congénita o adquirida a temprana edad. Por lo general, se presenta como una lesión única en forma de placa de aspecto vascular y, en ocasiones, con hipertricosis e hiperhidrosis (Fig. 3F).
Genética. Se han descrito mutaciones somáticas activadoras en GNA14 c.614A>T (p.Gln205Leu) en algunos casos de AP y de hemangioendoteliomas kaposiformes (HEK). Probablemente, ambos tumores forman parte del mismo espectro, siendo el AP el polo más benigno.
Curso clínico. Su historia natural es impredecible. Algunos casos se resuelven de 6 a 24 meses, dejando secuelas cutáneas mínimas, mientras que otros persisten en el tiempo, convirtiéndose en tumores más sólidos. Pueden complicarse con el fenómeno de Kasabach-Merritt (v. más adelante).
Diagnóstico. El diagnóstico se sospecha con la clínica, pero requiere confirmación histológica. La histología muestra numerosos capilares distribuidos a manera de nódulos que semejan “bolas de cañón” o “penachos”. La diferenciación entre el AP y el HEK más superficial es muchas veces es imposible. Son negativos para GLUT-1 y positivos para WT-1, D2-40, PROX1, CD31 y CD34.
Diagnóstico diferencial. Deben ser diferenciados de los miofibromas infantiles, del dermatofibrosarcoma protuberans congénito y de otros tumores, así como de malformaciones vasculares.
Tratamiento. Los tratamientos con corticoides sistémicos, aspirina, propranolol y rapamicina tópica, tienen eficacias variables (v. complicaciones AP y HEK). La cirugía es una opción en determinados casos. Con o sin FKM, la rapamicina sistémica parece ser efectiva(17).
Granuloma piógeno
El granuloma piógeno es muy frecuente en niños, puede aparecer después de una herida o picadura, y se caracteriza por crecer rápidamente y ocasionar sangrado con el mínimo roce.
El granuloma piógeno (GP) o hemangioma lobular capilar es el segundo tumor vascular más frecuente en niños. Si bien, se puede manifestar a cualquier edad, son más frecuentes en la infancia. Sus características clínicas se exponen en las figuras 3G y H.
Genética. Se han detectado mutaciones en BRAF, NRAS, GNAQ y otros genes que activan la vía de señalización de la MAPK/ERK.
Diagnóstico. El diagnóstico es clínico. La histopatología es muy característica, con una proliferación lobular de vasos capilares separados por septos fibrosos.
Diagnóstico diferencial. Los GP se deben diferenciar del HI. Pueden confundirse con el nevus de Spitz o con la leishmaniasis cutánea.
Complicaciones. Se pueden ulcerar y sangrar. Son raros los reportes de presentaciones congénitas atípicas de GP diseminados con compromiso visceral(18).
Tratamiento. La mayoría de lesiones son pequeñas y se pueden resecar mediante afeitado con curetaje de la base o bien tratar con láser de colorante pulsado, Nd:YAG o CO2. El timolol tópico puede ser efectivo en algunos casos.
Tumores vasculares localmente agresivos
Hemangioendotelioma kaposiforme (HEK)
El hemangioendotelioma kaposiforme representa el tumor vascular localmente agresivo más frecuente en niños y puede complicarse con un fenómeno de Kasabach-Merritt, que ponga en riesgo la vida del paciente.
El HEK es una neoplasia rara que puede estar presente, tanto en la piel (Fig. 3I) como en el hueso, el mediastino y el retroperitoneo. Comparte ciertas características clínicas e histopatológicas con el AP y está frecuentemente asociado con el FKM.
Epidemiología. Se estima que está presente en 0,9 de cada 100.000 nacidos vivos. No tiene predilección por sexo. El 93% de los HEK se dan en la infancia. El 60% de los casos se inicia en el primer mes de vida. Puede ser congénito(19).
Diagnóstico. Ante la sospecha clínica, requiere confirmación histológica. La anatomía patológica muestra una proliferación de células fusiformes que infiltran dermis y tejido celular subcutáneo. Su extensión puede llegar hasta fascia y el músculo. Es un tumor GLUT-1 negativo. Al igual que el AP, es positivo para D2-40 y PROX1.
Complicaciones de AP y HEK. En la infancia, algunos AP y HEK pueden desarrollar una coagulopatía de curso agresivo, caracterizada por trombocitopenia grave por: atrapamiento plaquetario, hipofibrinogenemía de consumo y anemia hemolítica microangiopática, que se conoce como FKM. Esta complicación, que ocurre en el 71% de los casos de HEK, es más común en tumores de gran tamaño y retroperitoneales. Se debe sospechar ante el incremento súbito del tamaño tumoral y la aparición de petequias, y equimosis. El tratamiento consiste en medidas de soporte. La transfusión de concentrados de plaquetas debe evitarse por la acción angiogénica de los factores de crecimiento plaquetarios. El uso de corticoides es discutible. Las mejores respuestas terapéuticas se reportan con: rapamicina, vincristina, antiagregantes e interferón a-2a y 2b, que ya no se utiliza por riesgo de paraparesia espástica en niños pequeños(19).
Tratamiento. Algunos HEK disminuyen de tamaño, pero la resolución completa es poco común. La cirugía es el tratamiento de elección cuando sea posible. La rapamicina sistémica puede ser una buena alternativa terapéutica(17).
Malformaciones vasculares
Las malformaciones vasculares son anomalías del desarrollo, por lo que suelen estar presentes desde el nacimiento y persisten hasta la vida adulta. Se clasifican en: simples, combinadas, de grandes vasos y asociadas a otras anomalías.
Las malformaciones vasculares son el resultado de errores en la morfogénesis embrionaria de: capilares, venas, arterias y vasos linfáticos (Tabla IV).

La apariencia clínica, la historia natural, las manifestaciones asociadas y el abordaje diagnóstico y terapéutico dependen del tipo de vaso afectado y del compromiso cutáneo o extracutáneo de la anomalía. Son lesiones que permanecen toda la vida y con el tiempo pueden empeorar.
El reciente descubrimiento de los genes incluidos en la tabla IV y de las diferentes vías de señalización que intervienen en su desarrollo, ha permitido entender con mayor claridad su naturaleza, así como la posibilidad de desarrollar terapias dirigidas para tratar entidades que se creían incurables.
Malformaciones capilares
Las malformaciones capilares son un motivo de consulta muy frecuente en dermatología pediátrica. Las manchas en vino de Oporto y las manchas salmón son las malformaciones capilares más frecuentes, pero no las únicas. Se pueden tratar con láseres vasculares.
Las malformaciones capilares (MC) son un motivo de consulta muy frecuente. Representan vasos capilares hamartomatosos presentes desde el nacimiento y que persisten hasta la vida adulta. Si bien, las MC más conocidas son las manchas en vino de Oporto (MVO) y las manchas salmón (MS), estas no son las únicas que existen. Esta variabilidad ha sido reconocida por la ISSVA, que las clasifica en 7 categorías clínicas incluidas en la tabla IV. Esta clasificación tiene interés, porque permite establecer un pronóstico y la necesidad de realizar exploraciones complementarias.
Nevus simple
El nevus simple es la malformación capilar congénita más frecuente; ha recibido diferentes nombres ilustrativos como: mancha salmón (MS), picotazo de la cigüeña, beso del ángel, eritema nucal o mancha en alas de mariposa.
La MS es la malformación capilar más frecuente y afecta hasta al 82% de los recién nacidos. De manera característica, desaparecen parcialmente a la vitropresión y se hacen más evidentes con el llanto o aumentos de la temperatura. Los hallazgos clínicos y las localizaciones características se describen en las figuras 4A y B.

Figura 4. Manifestaciones clínicas de las malformaciones capilares. A. Mancha salmón: se aprecia como una mácula rosada de bordes mal definidos. Se localiza con frecuencia en: párpados, en el centro de la frente, filtrum nasal, alrededor de las alas nasales, vertex, nuca y espalda, particularmente en la región lumbosacra, cuando se presenta de forma aislada, es un marcador de bajo riesgo para disrafismo; B. Mancha en vino de Oporto: son manchas sólidas, de forma y tamaño variables. El color oscila entre rosa y rojo azulado, y pueden aparecer en cualquier localización; C. Malformación capilar reticular: se presenta como una mácula eritematoviolácea de aspecto parcheado o reticulado con bordes imprecisos y tenues. Pueden ocupar territorios extensos como en este caso, que se distribuye en el hemitórax izquierdo y extremidad ipsilateral; D. Malformación capilar reticular: En esta imagen, se distribuye de manera extensa en la extremidad inferior derecha sin sobrecrecimiento asociado; E. Malformación capilar reticular: En esta imagen se distribuye de manera extensa en la extremidad inferior derecha sin sobrecrecimiento asociado; F. Malformación capilar difusa con sobrecrecimiento (DCMO, siglas correspondientes en inglés a Diffuse Capillary Malformation with Overgrowth): Además de la malformación capilar reticulada los pacientes cursan con sobrecrecimiento de tejidos blandos y huesos. La mayoría tienen únicamente sobrecrecimiento en una extremidad; G Malformación capilar asociada con hipocrecimiento (CMU, siglas correspondientes en inglés a Capillary Malformation with Undergrowth): En contraste al DCMO la MCR se asocia a hipocrecimiento de la extremidad afectada; H. Malformación capilar geográfica: máculas o placas de color vino tinto o violáceas que se describen como: “geográficas”, porque tienen bordes muy bien delimitados e irregulares que remedan un mapa. Hay vesículas linfáticas en la superficie. Este paciente tiene además alteraciones del sistema venoso, que se aprecian en la fotografía, y sobrecrecimiento (síndrome de Klippel-Trénaunay); I. Malformación capilar- malformación arteriovenosa: se ven como máculas de color rosado pálido o parduzco con formas redondeadas u ovaladas y diámetros diversos. A menudo, hay alguna lesión de mayor tamaño que suele estar más caliente a la palpación, como se ve en el hemitórax superior derecho de esta paciente. Es característica la presencia de un halo blanquecino periférico (manchas de Bier); J. Cutis marmorata telangiectásica congénita: se caracteriza por la presencia de máculas reticuladas azuladas que remedan una livedo reticularis con algún grado de atrofia focal sobre las máculas. Se localizan, por lo general, en extremidades inferiores y en tronco; K. Cutis marmorata telangiectásica congénita: Las áreas de atrofia pueden persistir por muchos años (flecha amarilla). Imagen tomada dos meses después de la figura 4H. Es evidente, como el reticulado vascular resolvió pronto, lo que es característico en la cutis marmorata telangiectásica congénita. Las áreas de atrofia pueden persistir por muchos años; L. Telangiectasia: se ven como pequeños puntos rojos conformados por vasos capilares dilatados, que remedan una corona radiada. Pueden ser únicas o múltiples. M. Telangiectasias en la enfermedad de Rendu-Osler-Weber: las lesiones suelen distribuirse en la cara, labios y mucosa oral (como en el paciente ilustrado), lengua y dedos de las manos.
Curso clínico. Con el tiempo, suelen aclararse por sí solas y dejar de ser visibles. Las de la nuca suelen persistir.
Diagnóstico. El diagnóstico es generalmente clínico y, sobre todo, por la localización característica de las mismas. Aunque sean extensas, no requieren estudios complementarios.
Diagnóstico diferencial. Puede confundirse con las MC tipo MVO; no obstante, estas clínicamente están mejor delimitadas y, cuando se localizan en la cara, suelen estar lateralizadas.
Asociaciones. Las MS, aun siendo extensas, no se asocian a ningún síndrome en particular. En la región lumbosacra, se consideran un marcador de bajo riesgo para disrafismo. Si no se encuentran en relación con otro marcador, como un área de hipertricosis, desviación del pliegue, foseta sacra alta o lipoma, no obligan a realizar exploraciones complementarias.
Mancha en vino de Oporto
Las manchas en vino de Oporto (MVO) son fácilmente reconocibles por sus características clínicas y por su persistencia. Se pueden confundir con las manchas salmón y pueden asociarse con el síndrome de Sturge-Weber.
Son el segundo tipo de malformación capilar más frecuente. Su incidencia es de 3 por 1.000 recién nacidos(20,21). Sus características clínicas se exponen en la figura 4C.
Curso clínico. La localización determina el riesgo de las alteraciones asociadas y el comportamiento de las mismas. A diferencia de las manchas salmón, son persistentes. Las MVO tienden a oscurecerse y a volverse más gruesas, especialmente en la cara, donde pueden ocasionar sobrecrecimiento leve de la región afecta. En ocasiones, también pueden desarrollar granulomas piógenos en su superficie (Fig. 3G).
Diagnóstico. El diagnóstico de las MVO es generalmente clínico. Se sabe que son debidas a mutaciones somáticas en GNAQ.
Diagnóstico diferencial. Se pueden confundir con las manchas salmón o con los hemangiomas infantiles abortivos, o de proliferación mínima.
Asociaciones. Hasta en el 10%, las MVO faciales se pueden asociar con: angioma coroideo, glaucoma y/o angiomatosis leptomeningea, que es lo que se conoce como síndrome de Sturge-Weber (SSW). No siempre está presente la tríada cutánea, ocular y del SNC; hay quien también denomina SSW, cuando solo existen dos manifestaciones. El riesgo de SSW depende de la localización y extensión de la MVO. Las manchas que tienen mayor riesgo de asociarse a un SSW son las extensas, las bilaterales y las que se distribuyen en el área que va desde el canto externo del ojo hasta la región frontal incluida(22-24).
El glaucoma es la complicación ocular más frecuente. Está presente en el 10 al 15% de niños con MVO periocular y en hasta el 70% de los niños que, además, tienen angiomatosis leptomeningea. Las MVO con riesgo de glaucoma son las que se localizan en la región frontal y, especialmente, en el área periocular. Si bien, el párpado inferior no se encuentra incluido en esta localización, en un estudio reciente se señala que las MVO que afectan al párpado inferior, aun sin afectación frontal, pueden tener más riesgo de glaucoma(25).
La angiomatosis leptomeningea, cuando da síntomas, se manifiesta en forma de: convulsiones, déficits neurológicos focales, cefaleas y problemas cognitivos, probablemente secundarios al mal control de las crisis epilépticas.
Seguimiento. Los niños con MVO distribuidas en la región frontal y periocular, deben seguir controles oftalmológicos y neurológicos. Las MVO con afectación de párpado inferior, aun sin afectación frontal, deberían también seguir controles oftalmológicos. Los controles oftalmológicos deben ser de por vida, porque el glaucoma puede ser congénito o de instauración, incluso en la edad adulta. La necesidad de realizar pruebas de neuroimagen en pacientes asintomáticos y el mejor momento para realizarla, es objeto de controversia. Si bien, existe algún falso negativo en las resonancias con contraste realizadas antes de los 3 meses de edad, en manos de neuroradiólogos expertos se suelen reconocer signos radiológicos directos o, al menos, algún signo indirecto. De esta manera, es posible educar a los padres en cómo actuar en caso de una primera convulsión e incluso plantear tratamiento presintomático con antiepilépticos o rapamicina en casos con angiomatosis leptomeningea extensa(26).
Malformación capilar reticulada
Las malformaciones capilares reticuladas tienen un aspecto menos sólido que las manchas en vino de Oporto y se pueden asociar a sobrecrecimiento de las extremidades; por lo que, a menudo, se diagnostican erróneamente como síndrome de Klippel-Trénaunay.
La malformación capilar reticulada (MCR) es una MC que tiene aspecto clínico parcheado o reticulado, y se puede ver en tres escenarios clínicos: 1) de forma aislada (Figs. 4D y E); 2) Asociada a sobrecrecimiento de partes blandas (malformación capilar con sobrecrecimiento, DCMO por sus siglas en inglés) (Fig. 4F); y 3) asociada a macrocefalia (malformación capilar-macrocefalia/megalencefalia, MC-M) (Figs. 5A y B).

Figura 5. Síndromes asociados a sobrecrecimiento que cursan con malformaciones capilares.
A y B. Síndrome MC-M (malformación capilar-macrocefalia/megalencefalia): se caracteriza por la presencia de macrocefalia/megalencefalia, mancha salmón prominente en el philtrum o glabela, y malformación reticulada extensa en el tronco y extremidades; C y D. Síndrome CLAPO: se asocia a malformación capilar geográfica en la parte inferior de la cara. Consiste en una malformación capilar de labio inferior, malformación linfática de la cara y cuello, asimetría facial y sobrecrecimiento parcial o generalizado; E y F. Síndrome CLOVES: se asocia a malformación capilar geográfica, masas lipomatosas subyacentes, nevus epidérmicos y anormalidades esqueléticas. Los pacientes con CLOVES no tienen el nevus de tejido conectivo plantar (figura 5, G); ni el crecimiento tan progresivo y distorsionante del síndrome de Proteus (figura 5, H); I. Síndrome Klippel-Trénaunay: cursan con malformación capilar reticulada asociada a: sobrecrecimiento, alteraciones venosas y linfáticas. A diferencia de la malformación capilar con sobrecrecimiento (DCMO), la hipertrofia es de carácter progresivo.
Recientemente, se ha descrito una cuarta variable clínica de MCR asociada a hipocrecimiento (CMU por sus siglas en inglés) (Fig. 4G)(27). La mayoría de MCR son por mutaciones en PIK3CA o GNA11.
Diagnóstico. El diagnóstico de la MCR aislada es clínico. En caso de dismetría, se recomienda realizar radiografías telemétricas al iniciar la deambulación y control por un ortopeda. En caso de macrocefalia asociada, debe realizarse una resonancia magnética cerebral.
Diagnóstico diferencial. Se pueden confundir con el cutis marmorata telangiectásica congénita (CMTC), no obstante las MCR son más parcheadas, no tienen la atrofia característica ni son tan prominentes o violáceas como el CMTC.
Asociaciones. Las MCR pueden asociar un sobrecrecimiento de la extremidad afectada, así como alteraciones digitales de los pies en forma de sindactilia o dedo en sandalia. En raras ocasiones, compromete la extremidad contralateral o incluso a todo el hemicuerpo. Las MCR extensas con sobrecrecimiento (DCMO) no muestran un crecimiento acelerado ni tienen las complicaciones tromboembólicas del síndrome Klippel-Trénaunay (SKT), pues carecen del componente venoso y linfático. Tienen, por tanto, un pronóstico muy benigno (Tabla V)(28).

Las MCR también pueden ser parte del síndrome de malformación capilar-macrocefalia / megalencefalia (MC-M). Se caracteriza por la presencia de: MCR extensa en tronco y extremidades, mancha salmón prominente en el labio superior, philtrum o glabela y macrocefalia/megalencefalia. Los pacientes tienen una facies característica, con una frente prominente y un crecimiento asimétrico, que ya es evidente en los primeros meses de vida. Desde el punto de vista neurológico, presentan una asimetría cerebral con megalencefalia, ventriculomegalia y no es infrecuente que presenten: displasia cortical, polimicrogiria, malformación de Arnold-Chiari o herniación cerebelosa. Son niños que suelen tener hipotonía y retraso en el desarrollo. Al igual que ocurre en otras hemihipertrofias, se han reportado dos casos de tumor de Wilms asociados a MC-M (Tabla V)(29).
Seguimiento. Los pacientes con una MCR aislada o que solo presentan un sobrecrecimiento del tronco y extremidades requieren seguimiento clínico periódico. La mayoría de asimetrías son leves y no tienen impacto funcional. Los que tienen un sobrecrecimiento más pronunciado necesitan un seguimiento cercano por un ortopeda.
Los pacientes con DCMO no parecen tener un riesgo aumentado de tumor de Wilms. Los pacientes con MC-M requieren un manejo multidisciplinar. Al momento de la sospecha diagnóstica, se debe realizar una resonancia magnética craneal basal y se recomienda repetirla cada 6 meses hasta cumplir 2 años, y después cada 3 a 4 años.
Los casos reportados con hipocrecimiento son de curso benigno no progresivo. Se recomienda, al menos, un seguimiento clínico periódico, porque se desconocen otras posibles asociaciones a largo plazo(27).
Malformación capilar geográfica
Los pacientes con una malformación capilar geográfica, a menudo, tienen un sobrecrecimiento asociado, alteraciones venosas y alteraciones linfáticas, formando la tríada del síndrome de Klippel-Trénaunay.
La malformación capilar geográfica (MCG) es una variante de MC, de aspecto clínico diferente a la MVO o MCR. Se trata de máculas oscuras muy bien delimitadas, de forma geográfica y que, con el tiempo, suelen desarrollar vesículas linfáticas en su superficie, por lo que probablemente representen malformaciones capilares linfáticas en las que, en un momento inicial, no se observa el componente linfático (Fig. 4H).
Curso clínico. Con el tiempo, pueden desarrollar vesículas linfáticas y/o hemorrágicas en su superficie.
Diagnóstico. El diagnóstico de la MCG aislada es clínico.
Diagnóstico diferencial. Se pueden confundir con las MVO y, muchas veces, hasta que no aparecen vesículas linfáticas, puede ser imposible diferenciarlas. Las malformaciones venosas verrugosas (MVV) pueden tener un aspecto similar a las MCG en los recién nacidos y lactantes. Se diferencian, porque las MVV tienen hiperqueratosis y no cursan con sobrecrecimiento(30).
Asociaciones. Las MCG, a menudo, tienen sobrecrecimiento asociado, alteraciones venosas y linfáticas, formando la triada del SKT. A diferencia del DCMO, la hipertrofia es de carácter progresivo. La malformación venosa del SKT generalmente se manifiesta como: varicosidades, alteraciones del sistema venoso profundo y, con el tiempo, puede hacerse visible una vena anómala en la cara lateral de la extremidad afectada, que representa la persistencia de la vena embrionaria o “vena lateral marginal”. Raras veces está presente desde el nacimiento. Estas alteraciones constituyen un riesgo para desarrollar trombosis venosa con riesgo de tromboembolismo pulmonar (Tabla V).
Las MCG también se pueden ver en otros síndromes asociados a sobrecrecimiento, como el síndrome CLAPO, CLOVES y Proteus (Figs. 5C-I)(31,32). Es importante reconocer los diferentes síndromes de sobrecrecimiento asociados a malformaciones capilares, porque las comorbilidades asociadas y el pronóstico difieren entre ellos (Tabla V).
Seguimiento. Los pacientes con MCG en las extremidades deben ser evaluados periódicamente para detectar dismetrías. Se recomienda una valoración ortopédica entre el primer año y segundo de vida, en caso de dismetrías importantes. No hay consenso sobre las exploraciones complementarias o a qué edad solicitarlas en estos pacientes. En nuestra experiencia, resulta útil realizar en algún momento, una resonancia magnética con contraste para descartar la presencia de alteraciones venosas asociadas y determinar el alcance del sobrecrecimiento. Por otro lado, de existir alteraciones venosas, hay que considerar la administración de heparina en situaciones de riesgo de trombosis.
Malformación capilar – Malformación arteriovenosa
Las malformaciones capilares asociadas a este síndrome son: pequeñas, múltiples, de aparición progresiva y familiar. Es importante reconocerlas, porque estos pacientes pueden asociar malformaciones arteriovenosas periféricas y en el sistema nervioso central.
El síndrome de malformación capilar – malformación arteriovenosa (MC-MAV) es un trastorno con patrón de herencia autosómica dominante resultado de mutaciones inactivadoras del gen RASA1 que codifica para el activador 1 de la proteína Ras p21. Es importante identificarlo, porque hasta un tercio de los pacientes tiene una malformación arteriovenosa asociada (MAV) o fístula arteriovenosa (FAV) en el sistema nervioso central o periférico. En una minoría de los casos, puede haber mutaciones en el gen EPHB4, entidad que algunos autores denominan MC-MAV2.
Epidemiología. La prevalencia se estima en 0,85-1 por 100.000 nacidos(33,34).
Manifestaciones clínicas. Las malformaciones capilares pueden estar presentes al nacer, pero aparecen más con los años. Se trata de máculas de pequeño tamaño de coloración rosa pálido o pardo, a menudo, con un halo blanquecino en la periferia. Con frecuencia, hay una lesión de mayor tamaño que suele estar más caliente a la palpación. Algunos pacientes desarrollan telangiectasias en cara y extremidades. Estas pueden tener un halo de vasoconstricción (manchas de Bier) (Fig. 4I). La penetrancia de este síndrome es prácticamente completa.
Diagnóstico. El diagnóstico suele ser clínico. Sin antecedentes familiares, el aspecto clínico de las malformaciones permite sospechar el diagnóstico. En la ecografía Doppler puede detectarse, a veces, un flujo más elevado que en una simple malformación capilar.
Diagnóstico diferencial. El color pardo de estas MC puede hacerlas confundir con manchas café con leche. Las telangiectasias faciales, a veces, se confunden con la enfermedad de Rendu-Osler-Weber.
Asociaciones. Los pacientes con MC-MAV tienen un riesgo del 30% de tener una MAV de alto flujo en tejidos blandos, 9% en el cerebro y de un 2% en la médula espinal. Los afectados con MC-MAV parecen tener más riesgo de presentar MAV o FAV de alto flujo asociadas que los pacientes con MC-MAV2. Cuando la MAV está en extremidades, es lo que se conoce como síndrome de Parkes-Weber.
Seguimiento. El seguimiento de estos pacientes debe ser estrecho. Los autores recomiendan la obtención de una RM con contraste craneal y espinal, cuando se identifica el cuadro clínico característico, puesto que la detección precoz permite realizar un tratamiento antes de que debute con una complicación hemorrágica(33). No está claro si debe repetirse en la edad adulta.
Cutis marmorata telangiectásica congénita
El cutis marmorata telangiectásica congénita es una variedad de malformación capilar de curso benigno, que no suele estar asociado con otras alteraciones.
El CMTC, también conocido como flebectasia congénita, es una anomalía vascular rara, que se caracteriza por la presencia de máculas reticuladas violáceas y atróficas que remedan una livedo reticularis (Figs. 4J y K)(35).
Curso clínico. Con el tiempo, las lesiones suelen hacerse menos visibles y pueden llegar a ser casi imperceptibles. En algunas ocasiones, se puede observar hipotrofia leve en la extremidad comprometida, pero mejora con la edad por sí sola.
Diagnóstico. El diagnóstico es clínico y no requiere exploraciones complementarias. Corresponde a una lesión de bajo flujo, por lo que el Doppler no suele ser de utilidad.
Diagnóstico diferencial. El diagnóstico diferencial del CMTC incluye: cutis marmorata fisiológico del recién nacido; los hemangiomas abortivos o de proliferación mínima; las malformaciones capilares reticuladas y algunas lesiones de lupus eritematoso neonatal (LEN).
Asociaciones. La asociación del CMTC localizado con otras alteraciones probablemente es casual. La única asociación que podría ser real es con glaucoma en los CMTC faciales. Las formas generalizadas pueden formar parte del síndrome de Adams Oliver con aplasia cutis, defectos digitales y cardiopatía congénita(35).
Seguimiento. Los pacientes tienen un pronóstico excelente. No requieren de seguimiento estrecho. En los casos de CMTC facial, es recomendable una exploración oftalmológica(35).
Telangiectasias
Las telangiectasias de distribución generalizada no siempre están asociadas a la telangiectasia hemorrágica hereditaria, hay entidades asintomáticas y con curso igual de benigno que también las presentan.
Las telangiectasias se ven como una pápula de aspecto vascular central de la que irradian capilares dilatados y de aquí el nombre de araña vascular (Fig. 4L). Pueden ser únicas o múltiples.
Se han descrito dos formas de telangiectasias múltiples:
1. Telangiectasia nevoide unilateral (TNU). Hace referencia a una alteración esporádica en la que existen múltiples telangiectasias de distribución lineal o segmentaria. Con frecuencia, se localizan en el cuello o en extremidades superiores.
2. Telangiectasia benigna hereditaria (TBH). La TBH es un trastorno con patrón de herencia autosómico dominante con telangiectasias de distribución generalizada. Las lesiones aparecen en edad temprana, generalmente en la cara y, con el tiempo, se van generalizando por diferentes localizaciones.
Diagnóstico diferencial. En pacientes con telangiectasias múltiples, debe plantearse el diagnóstico diferencial con la telangiectasia hemorrágica hereditaria (THH) o enfermedad de Rendu-Osler-Weber. La THH es una enfermedad de herencia autosómica dominante que requiere un manejo multidisciplinario. Puede debutar con: epistaxis, sangrados intestinales o de otras vísceras. Clínicamente, las telangiectasias de la THH suelen adquirirse en la adolescencia o edad adulta, no parecen arañas vasculares, sino que son lesiones tipo mácula o pápula de color rojo intenso y de mayor tamaño. Las lesiones afectan de forma característica a: cara, labios, mucosa oral, lengua y dedos de las manos (Fig. 4M). Se deben hacer pruebas de imagen en estos pacientes, para descartar malformaciones arteriovenosas cerebrales, pulmonares y hepáticas, que determinan el pronóstico. Se cree que algunos pacientes con TBH pueden tener, en realidad, un síndrome de MC-MAV, ya que en esta entidad, también aparecen telangiectasias; no obstante, en la mayoría de casos, es posible observar alguna MC característica.
Asociaciones. Tanto la TNU como la TBH no se asocian a otras alteraciones. Son entidades benignas y de buen pronóstico, y no es necesario realizar exploraciones complementarias.
Tratamiento. Las telangiectasias pueden tratarse con láser de colorante pulsado o de Nd:YAG.
Malformaciones venosas
Las malformaciones venosas son alteraciones en la morfogénesis de las venas. Son malformaciones vasculares de bajo flujo, que pueden producir dolor en relación a fenómenos de trombosis.
Las malformaciones venosas (MV) son anomalías vasculares no proliferativas, de bajo flujo, compuestas por canales venosos ectáticos anómalos. Generalmente, están presentes desde el nacimiento, sin embargo, pueden no hacerse evidentes con el crecimiento secundario a la dilatación venosa gradual. Si bien, en la clasificación de la ISSVA se describen 7 malformaciones venosas simples (Tabla IV), en este artículo se revisaran solo algunas.
Las MV esporádicas son el resultado de mutaciones somáticas activadoras en el gen TEK y en PIK3CA (50% y 25% respectivamente) y se identifican, clínicamente, como masas o nódulos de: tonalidad azul, blandas, compresibles y de tamaño variable en piel, mucosas o intramusculares (Figs. 6A y B). La forma familiar es de herencia autosómica dominante, por mutación en línea germinal de TEK y se presenta con múltiples lesiones en piel y mucosas. Puede haber afectación de órganos internos.

Figura 6. Características clínicas de las malformaciones venosas y linfáticas.
A. Malformación venosa: las malformaciones venosas comunes se identifican clínicamente como masas o nódulos de tonalidad azul, blandas, compresibles y de tamaño variable. Pueden ser: profundas, superficiales, solitarias, localizadas, segmentarias, difusas o múltiples; B: malformación venosa que afecta piel, lengua y mucosa oral en un lactante; C. Síndrome del nevus azul en tetina de goma (BRBNS): compromiso cutáneo multifocal de un paciente afecto por BRBNS. Se observan múltiples pápulas y nódulos azul violáceos con aspecto de “tetina de goma”; D: laparotomía exploratoria del paciente ilustrado en la figura 6C, donde se evidencian múltiples malformaciones venosas intestinales; E. Malformación glomovenosa (MGV): se puede presentar desde una pequeña lesión nodular solitaria azul-violácea, hasta formas extensas conformadas por placas de distribución segmentaria; F. Malformación venosa verrugosa: placas rojo violáceas de bordes irregulares y de grosor variable; G. Malformación linfática: las malformaciones linfáticas (ML) microquísticas pueden aparecer en piel y mucosas. Aparecen como vesículas de contenido seroso o hemorrágico, de 1 a 2 mm de diámetro con aspecto de “huevos de caviar”; H. ML macroquística: se observa como una tumoración de color piel que, a veces, es posible observar que transiluminan; I. Linfedema: se manifiesta como un incremento del volumen subcutáneo, bien delimitado, blando y renitente. Puede aparecer en cualquier parte del cuerpo, aunque usualmente se presenta en extremidades inferiores más que en superiores y, en algunos casos, hay compromiso genital; J – K. Malformación arteriovenosa (MAV): las MAV en etapas tempranas (fase 1 de Schöbinger) son difíciles de diferenciar de las manchas en vino de Oporto. En la fase 2, se ven como nódulos azul violáceos mal delimitados, con vasos venosos en su superficie y de consistencia firme. Al momento de palparlas, es característico sentir un pulso o “thrill”. En esta imagen, se aprecia una localización usual de las MAV en la cara de una adolescente; L: MAV en el dorso de pie de una mujer de 14 años en fase 2 de Schöbinger, con cambios de dermatosis purpúrica pigmentaria; M: MAV de la paciente anterior en progresión a fase 3 con necrosis y ulceración.
Epidemiología. Las MV comunes y familiares representan el 95% del total de MV. Tienen una prevalencia del 1% y una incidencia estimada en 1-2 por 10.000 nacidos vivos.
Diagnóstico. El diagnóstico de malformación venosa es clínico. La ecografía Doppler confirmará que se trata de una lesión de flujo lento. Puede detectar también flebolitos calcificados. La resonancia magnética es útil para definir la extensión y para planear tratamiento(21).
Diagnóstico diferencial. Debe establecerse con hemangiomas profundos u otros tumores vasculares profundos.
Asociaciones y complicaciones. Las asociaciones y complicaciones dependerán de la localización y del tamaño. Las MV cefálicas con frecuencia presentan compromiso cosmético y funcional. Característicamente, aumentan de tamaño con las maniobras de Valsalva. Puede extenderse a planos profundos como: músculo, fosa infratemporal y orbitaria. En pacientes con MV en la lengua o vía aérea, puede asociarse al síndrome de apnea obstructiva del sueño. Las MV cefálicas pueden tener anomalías del drenaje venoso intracraneal, pero no se asocian a MV intracerebrales.
Las MV localizadas en tronco y extremidades pueden infiltrar estructuras más profundas como: músculo, hueso y articulaciones, que pueden condicionar dolor con el ejercicio y episodios de dolor matutino. En los casos de infiltración articular, puede determinar hemartrosis con riesgo de degeneración del cartílago articular. Las opciones terapéuticas se exponen en la tabla VI.
La mayoría de MV grandes, al ser anomalías con flujo lento, tienen predisposición a desarrollar episodios de coagulopatía intravascular localizada y trombosis, con elevación del dímero D y disminución del fibrinógeno(36). Las opciones terapéuticas se exponen en la tabla VI.

Síndrome del nevus azul en tetina de goma (“Blue Rubber Bleb Nevus Syndrome”, BRBNS)
Este síndrome esporádico se caracteriza por la presencia de múltiples pápulas de color vinoso o azulado, y de tacto que asemeja a una “tetina de goma”. A veces, tiene una lesión “predominante” de gran tamaño que, característicamente, muestra vasos rojizos superficiales con un aspecto que remeda a un helecho. Tienen compromiso cutáneo multifocal (Fig. 6C) y del tracto digestivo (Fig. 6D), lo que provoca sangrados continuos y anemia ferropénica(37,38).
Malformación glomovenosa (MGV)
Representa una variante de MV poco frecuente. Son el resultado de mutaciones germinales en heterocigosis del gen de la glomulina (GLMN). Su apariencia clínica se aprecia en la figura 6E. Con frecuencia, son dolorosas al tacto, parcialmente comprimibles y con un aspecto “empedrado”. Suelen tener una distribución segmentaria. A diferencia de las MV, no se modifican con las maniobras de Valsalva o con la dependencia. Son más comunes en tronco y en extremidades. No suelen afectar órganos internos o articulaciones, y el compromiso de mucosa oral es excepcional. La histopatología es muy útil para el diagnóstico, ya que se evidencian dilataciones vasculares rodeadas de células glómicas y hallazgos anatomopatológicos típicos de las MGV(39).
Malformación venosa verrugosa (MVV)
Conocida previamente como hemangioma verrugoso. Se clasifica como un tipo superficial de malformación venosa y, recientemente, se ha descrito que son el resultado de mutaciones somáticas en el gen MAP3K3. Se caracterizan, clínicamente, por ser placas rojo violáceas de bordes irregulares y de grosor variable (Fig. 6F). Con los años, desarrollan hiperqueratosis en la superficie. Por lo general, tienen distribución lineal, aunque también hay formas parcheadas y unilaterales. Pueden cursar con dolor y sangrado superficial. El diagnóstico diferencial debe establecerse con el angioqueratoma. El tratamiento de elección en lesiones circunscritas es la exéresis completa (Tabla VI)(30).
Otras anomalías vasculares provisionalmente no clasificadas
Anomalía vascular fibroadiposa (FAVA)
Se trata de un tipo de malformación de bajo flujo que afecta el músculo y que se caracteriza por la presencia de canales venosos anormales entre el tejido adiposo. No suele ser aparente a simple vista o puede provocar una distorsión del contorno de la región afectada. Generalmente, se presenta en el músculo gastrocnemius y debuta con dolor en la pantorrilla y contractura muscular. En algunos casos, se han detectado mutaciones somáticas en PIK3CA. El tratamiento se expone en la tabla VI.
Malformaciones linfáticas
Las malformaciones linfáticas pueden tener aumento súbito de tamaño secundario a procesos infecciosos; en estos casos, es necesario el manejo con antibióticos sistémicos y con antiinflamatorios no esteroideos (AINE).
Las malformaciones linfáticas (ML) son el resultado de anomalías del desarrollo del sistema linfático que condicionan un drenaje anómalo de la linfa debido a: aplasia, hipoplasia o disrupción de los canales linfáticos. Con frecuencia, están presentes desde el nacimiento, pero pueden manifestarse años más tarde. Su espectro clínico es amplio (Figs. 6G-I), y se clasifican en: ML comunes, anomalía linfática generalizada, anomalías del conducto torácico y linfedema (Tabla IV). En este artículo, se revisaran solo las ML comunes.
Malformaciones linfáticas comunes
Se trata de dilataciones anormales de los canales linfáticos. Dependiendo del tamaño de las cavidades, se clasifican en: macroquísticas (cavidades mayores a 2 cm), microquísticas (menores de 2 cm) o mixtas. Las macroquísticas son más frecuentes en el cuello y región torácica y, a menudo, se detectan en las ecografías prenatales (Fig. 6H). Se presentan como tumoraciones color piel que transiluminan. Las microquísticas se presentan como múltiples vesículas agrupadas de contenido claro o hemorrágico. Pueden aparecer en cualquier localización (Fig. 6G).
Epidemiología. La incidencia de las ML en general, se estima en 1 a 2.000/4.000 nacidos vivos, sin predilección por raza o género(39).
Diagnóstico. El diagnóstico de las ML comunes es clínico. La eco-Doppler confirma la presencia de cavidades no compresibles, sin vascularización en su interior. Puede ser necesaria una RM para valorar la extensión en profundidad(40).
Complicaciones. Las ML macroquísticas, en ocasiones, aumentan súbitamente de tamaño, con signos inflamatorios y dolor. Estos episodios pueden estar provocados por sangrado intralesional o infección, y responden a antibioticoterapia y una tanda corta de corticoides orales. Estos episodios pueden provocar fibrosis con diminución del tamaño de las lesiones.
Tratamiento. El tratamiento depende del tipo y de la extensión de la ML (Tabla VI).
Malformaciones arteriovenosas
Las malformaciones arteriovenosas representan el grupo más peligroso de las anomalías vasculares. Pueden ulcerarse, provocar sangrados de difícil manejo, necrosis cutánea e insuficiencia cardiaca de gasto elevado.
Las malformaciones arteriovenosas son las menos frecuentes, pero constituyen el grupo más agresivo de las anomalías vasculares. Suelen estar presentes desde el nacimiento, pero el 30% se hacen evidentes en la adolescencia. Son el resultado de la conexión anormal entre los vasos arteriales y venosos sin un lecho capilar intermedio. Se caracterizan por ser lesiones de alto flujo y baja resistencia vascular periférica, y con frecuencia se asocian a fístulas. Se han descrito mutaciones somáticas en la vía de señalización de RAS-MAPK-ERK(41).
Desde el punto de vista clínico, suelen pasar por 4 estadios evolutivos conocidos como las fases de Schöbinger: 1) fase quiescente o asintomática, en la que simulan una malformación capilar y puede haber aumento de temperatura local; 2) fase de expansión, en la que la malformación crece e invade estructuras profundas; 3) fase destructiva, que se caracteriza por: ulceración, sangrado y necrosis; y 4) fase de descompensación, que se asocia a: insuficiencia cardíaca, hipertensión venosa o muerte. Los diferentes estadios clínicos se ilustran en las figuras 6J-M.
Epidemiología. Se estima que la incidencia es de 1 por 100.000 nacidos vivos.
Diagnóstico. La ecografía Doppler pone en evidencia un flujo alto, lo que las diferencia de las MV y ML. La resonancia magnética con contraste es útil para confirmar y establecer sus límites. La angiografía convencional o angioresonancia resulta imprescindible antes de abordar el tratamiento.
Tratamiento. El tratamiento de elección es la resección completa con o sin embolización previa en el mismo acto o unas horas antes. Lamentablemente, a menudo, esto no es posible. La ligadura proximal, embolización proximal o resecciones parciales están abocadas al fracaso y a complicaciones importantes (Tabla VI).
Conclusiones. Entre tanta nomenclatura tan diversa y confusa, las anomalías vasculares probablemente representan uno de los trastornos que más dificultades ha ocasionado para categorizar adecuadamente. Si bien, las clasificaciones son siempre una simplificación de la realidad, la clasificación de la ISSVA es útil para encuadrar estas anomalías. Reconocer las malformaciones vasculares más frecuentes en la infancia, ayuda a evitar, en la mayoría de los casos, estudios complementarios innecesarios. Identificar las asociaciones, favorece un mejor abordaje diagnóstico y establece parámetros más claros en el seguimiento de estos pacientes.
El rol del médico de Atención Primaria y del pediatra es muy importante en el enfoque inicial de las malformaciones vasculares por varias razones. Son los profesionales idóneos para filtrar las lesiones de curso benigno, que solo requieren seguimiento y no necesitan abordajes agresivos. Además, son los facultativos de primera línea que, por su estrecha relación con el ejercicio de promover la salud y prevenir la enfermedad, detectan los casos graves que requieren manejo prioritario.
Bibliografía
Los asteriscos muestran el interés del artículo a juicio de los autores.
1.*** ISSVA Classification of Vascular Anomalies ©2018 International Society for the Study of Vascular Anomalies. Disponible en: issva.org/classification.
2. Baselga Torres E, Bernabéu Wittel J, van Esso Arbolave DL, Febrer Bosch MI, Carrasco Sanz Á, de Lucas Laguna R, et al. Spanish consensus on infantile haemangioma. An Pediatr (Barc). 2016; 85: 256-65.
3. Steiner JE, Drolet BA. Classification of Vascular Anomalies: An Update. Semin Intervent Radiol. 2017; 34: 225-32.
4. Baselga E, Roe E, Coulie J, Muñoz FZ, Boon LM, McCuaig C, et al. Risk Factors for Degree and Type of Sequelae After Involution of Untreated Hemangiomas of Infancy. JAMA Dermatol. 2016; 152: 1239-43.
5.*** Luu M, Frieden IJ. Haemangioma: clinical course, complications and management. Br J Dermatol. 2013; 169: 20-30.
6. O’Brien KF, Shah SD, Pope E, Phillips RJ, Blei F, Baselga E, et al. Late growth of infantile hemangiomas in children >3 years of age: A retrospective study. J Am Acad Dermatol. 2019; 80: 493-9.
7. Iacobas I, Burrows PE, Frieden IJ, Liang MG, Mulliken JB, Mancini AJ, et al. LUMBAR: association between cutaneous infantile hemangiomas of the lower body and regional congenital anomalies. J Pediatr. 2010; 157: 795-801.e1-7.
8.*** Garzon MC, Epstein LG, Heyer GL, Frommelt PC, Orbach DB, Baylis AL, et al. PHACE Syndrome: Consensus-Derived Diagnosis and Care Recommendations. J Pediatr. 2016; 178: 24-33.e2.
9. Chang SJ, Yu W, Gu Y, Han Y, Shang Y, Chang L, et al. Location of infantile hemangioma is a predictor of volumetric sequelae after involution. J Dermatol. 2019; 46: 371-5.
10. Krowchuk DP, Frieden IJ, Mancini AJ, Darrow DH, Blei F, Greene AK, et al. Clinical Practice Guideline for the Management of Infantile Hemangiomas. Pediatrics; 2019. p. 143.
11. Frieden IJ, Püttgen KB, Drolet BA, Garzon MC, Chamlin SL, Pope E, et al. Management of infantile hemangiomas during the COVID pandemic. Pediatr Dermatol. 2020; 37: 412-8.
12. Püttgen K, Lucky A, Adams D, Pope E, McCuaig C, Powell J, et al. Topical Timolol Maleate Treatment of Infantile Hemangiomas. Pediatrics; 2016. p. 138.
13.*** Léauté-Labrèze C, Baselga Torres E, Weibel L, Boon LM, El Hachem M, van der Vleuten C, et al. The Infantile Hemangioma Referral Score: A Validated Tool for Physicians. Pediatrics; 2020. p. 145.
14. Nasseri E, Piram M, McCuaig CC, Kokta V, Dubois J, Powell J. Partially involuting congenital hemangiomas: a report of 8 cases and review of the literature. J Am Acad Dermatol. 2014; 70: 75-9.
15. Boix-Vilanova J, Baselga E, Vera A, González-Hermosa MDR, Azaña JM, Martín-Santiago A. Expanding the phenotypes of congenital hemangiomas. Pediatr Dermatol. 2020; 37: 872-6.
16. Ayturk UM, Couto JA, Hann S, Mulliken JB, Williams KL, Huang AY, et al. Somatic Activating Mutations in GNAQ and GNA11 Are Associated with Congenital Hemangioma. Am J Hum Genet. 2016; 98: 789-95.
17. Wang H, Guo X, Duan Y, Zheng B, Gao Y. Sirolimus as initial therapy for kaposiform hemangioendothelioma and tufted angioma. Pediatr Dermatol. 2018; 35: 635-8.
18. Alomari MH, Kozakewich HPW, Kerr CL, Uller W, Davis SL, Chaudry G, et al. Congenital Disseminated Pyogenic Granuloma: Characterization of an Aggressive Multisystemic Disorder. J Pediatr. 2020. DOI: 10.1016/j.jpeds.2020.06.079.
19. Croteau SE, Liang MG, Kozakewich HP, Alomari AI, Fishman SJ, Mulliken JB, et al. Kaposiform hemangioendothelioma: atypical features and risks of Kasabach-Merritt phenomenon in 107 referrals. J Pediatr. 2013; 162: 142-7.
20. Shirley MD, Tang H, Gallione CJ, Baugher JD, Frelin LP, Cohen B, et al. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med. 2013; 368: 1971-9.
21. Baselga E. Vascular Malformations. En: Bolognia J, Schaffer JV, Cerroni L. Dermatology. Fourth edition ed. Philadelphia: Elsevier; 2018. p. 1805-27.
22. Higueros E, Roe E, Granell E, Baselga E. Sturge-Weber Syndrome: A Review. Actas Dermosifiliogr. 2017; 108: 407-17.
23.*** Waelchli R, Aylett SE, Robinson K, Chong WK, Martínez AE, Kinsler VA. New vascular classification of port-wine stains: improving prediction of Sturge-Weber risk. Br J Dermatol. 2014; 171: 861-7.
24. Dutkiewicz AS, Ezzedine K, Mazereeuw-Hautier J, Lacour JP, Barbarot S, Vabres P, et al. A prospective study of risk for Sturge-Weber syndrome in children with upper facial port-wine stain. J Am Acad Dermatol. 2015; 72: 473-80.
25. Ha A, Kim JS, Baek SU, Park YJ, Jeoung JW, Park KH, et al. Facial Port-Wine Stain Phenotypes Associated with Glaucoma Risk in Neonates. Am J Ophthalmol. 2020; 220: 183-90.
26. Bar C, Pedespan JM, Boccara O, Garcelon N, Levy R, Grévent D, et al. Early magnetic resonance imaging to detect presymptomatic leptomeningeal angioma in children with suspected Sturge-Weber syndrome. Dev Med Child Neurol. 2020; 62: 227-33.
27. Cubiró X, Rozas-Muñoz E, Castel P, Roé Crespo E, García-Melendo C, Puig L, et al. Clinical and genetic evaluation of six children with diffuse capillary malformation and undergrowth. Pediatr Dermatol. 2020; 37: 833-8.
28.*** Lee MS, Liang MG, Mulliken JB. Diffuse capillary malformation with overgrowth: a clinical subtype of vascular anomalies with hypertrophy. J Am Acad Dermatol. 2013; 69: 589-94.
29. Peterman CM, Vadeboncoeur S, Mulliken JB, Fishman SJ, Liang MG. Wilms tumor screening in diffuse capillary malformation with overgrowth and macrocephaly-capillary malformation: A retrospective study. J Am Acad Dermatol. 2017; 77: 874-8.
30. Boccara O, Ariche-Maman S, Hadj-Rabia S, Chrétien-Marquet B, Frassati-Biaggi A, Zazurca F, et al. Verrucous hemangioma (also known as verrucous venous malformation): A vascular anomaly frequently misdiagnosed as a lymphatic malformation. Pediatr Dermatol. 2018; 35: e378-e81.
31. Martínez-López A, Blasco-Morente G, Pérez-López I, Herrera-García JD, Luque-Valenzuela M, Sánchez-Cano D, et al. CLOVES syndrome: review of a PIK3CA-related overgrowth spectrum (PROS). Clin Genet. 2017; 91: 14-21.
32. Rodríguez-Laguna L, Ibáñez K, Gordo G, García-Minaur S, Santos-Simarro F, Agra N, et al. CLAPO syndrome: identification of somatic activating PIK3CA mutations and delineation of the natural history and phenotype. Genet Med. 2018; 20: 882-9.
33. Valdivielso-Ramos M, Martín-Santiago A, Azaña JM, Hernández-Núñez A, Vera A, Pérez B, et al. Capillary malformation-arteriovenous malformation syndrome: a multicentre study. Clin Exp Dermatol. 2020.
34. Wooderchak-Donahue WL, Johnson P, McDonald J, Blei F, Berenstein A, Sorscher M, et al. Expanding the clinical and molecular findings in RASA1 capillary malformation-arteriovenous malformation. Eur J Hum Genet. 2018; 26: 1521-36.
35. Bui TNPT, Corap A, Bygum A. Cutis marmorata telangiectatica congenita: a literature review. Orphanet J Rare Dis. 2019; 14: 283.
36. Dompmartin A, Acher A, Thibon P, Tourbach S, Hermans C, Deneys V, et al. Association of localized intravascular coagulopathy with venous malformations. Arch Dermatol. 2008; 144: 873-7.
37. Ivars M, Martín-Santiago A, Baselga E, Guibaud L, López-Gutiérrez JC. Fern-shaped patch as a hallmark of blue rubber bleb nevus syndrome in neonatal venous malformations. Eur J Pediatr. 2018; 177: 1395-8.
38. Bolognia J, Schaffer JV, Cerroni L. Dermatology. Fourth edition ed. Philadelphia: Elsevier; 2018. p. 1805-27.
39. Pang C, Lim CS, Brookes J, Tsui J, Hamilton G. Emerging importance of molecular pathogenesis of vascular malformations in clinical practice and classifications. Vasc Med. 2020; 25: 364-77.
40. Sadick M, Müller-Wille R, Wildgruber M, Wohlgemuth WA. Vascular Anomalies (Part I): Classification and Diagnostics of Vascular Anomalies. Rofo. 2018; 190: 825-35.
41. Barbosa Do Prado L, Han C, Oh SP, Su H. Recent Advances in Basic Research for Brain Arteriovenous Malformation. Int J Mol Sci; 2019. p. 20.
42. Alonso San Pablo MT, Calderón-Castrat X. Anomalías vasculares. Pediatr Integral. 2016; XX(3): 159-68.
Bibliografía recomendada
- ISSVA Classification of Vascular Anomalies ©2018 International Society for the Study of Vascular Anomalies. Disponible en: issva.org/classification.
En esta página web aparece la clasificación más actualizada de las anomalías vasculares.
- Luu M, Frieden IJ. Haemangioma: clinical course, complications and management. Br J Dermatol. 2013; 169: 20-30.
Este es un artículo de revisión sobre los hemangiomas de la infancia, en el que se repasan formas de presentación, riesgo de asociaciones e indicaciones de tratamiento.
- Garzon MC, Epstein LG, Heyer GL, Frommelt PC, Orbach DB, Baylis AL, et al. PHACE Syndrome: Consensus-Derived Diagnosis and Care Recommendations. J Pediatr. 2016; 178: 24-33.e2.
Este es un artículo en el que se define el síndrome PHACE, se recogen todas las alteraciones que pueden verse en este síndrome y se señala el seguimiento indicado para cada paciente.
- Waelchli R, Aylett SE, Robinson K, Chong WK, Martínez AE, Kinsler VA. New vascular classification of port-wine stains: improving prediction of Sturge-Weber risk. Br J Dermatol. 2014; 171: 861-7.
En este artículo, se desmiente el concepto extendido de que las manchas en vino de Oporto de distribución en la primera rama del trigemino son las que tiene riesgo de SSW. Estos autores encuentran que la afectación de la frente es prácticamente lo único importante.
- Léauté-Labrèze C, Baselga Torres E, Weibel L, Boon LM, El Hachem M, van der Vleuten C, et al. The Infantile Hemangioma Referral Score: A Validated Tool for Physicians. Pediatrics; 2020. p. 145.
Este es un artículo muy útil para Atención Primaria, pues presenta un algoritmo sencillo que permite determinar los hemangiomas de mayor riesgo o en los que cabe considerar remisión al pediatra y/o dermatólogo pediatra.
- Lee MS, Liang MG, Mulliken JB. Diffuse capillary malformation with overgrowth: a clinical subtype of vascular anomalies with hypertrophy. J Am Acad Dermatol. 2013; 69: 589-94.
Estos autores describen una serie de pacientes con malformación capilar asociada a sobrecrecimiento. Es importante el reconocimiento de este fenotipo que, a menudo, se confunde con Klippel-Trénaunay cuando, en realidad, es un cuadro de mejor pronóstico.
| Caso clínico 1 |
|
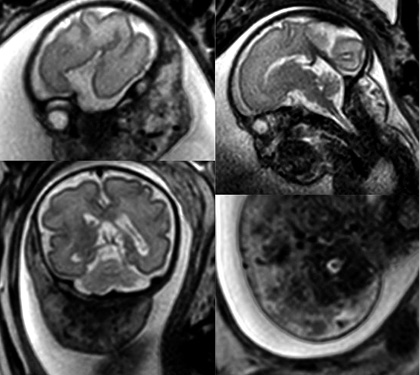
Primigestante de 32 años sin antecedentes patológicos de interés que, en la ecografía prenatal de las 20 semanas, se identifica en el feto una lesión tumoral de alto flujo localizada en región laterocervical. El embarazo transcurre sin incidencias hasta la semana 30 que presenta polihidramnios severo y se decide realizar resonancia fetal a la semana 32 y otra a la 34. La primera resonancia pone en evidencia una “lesión en bufanda” con vacío de flujo en T2 con dimensiones globales de 83 x 44 x 30 mm (Fig. 7).
Figura 7. Secuencias T2 “lesión en bufanda”. Vacío de señal central. En la segunda, se aprecia una lesión con diámetros similares con prominencia de algunas ramas de la arteria carótida externa e ingurgitación de la vena yugular ipsilateral. Con estos hallazgos, se sospecha una anomalía vascular de alto flujo, probablemente compatible con hemangioma. Dentro de los diferenciales, se contemplaron HI, HC, MAV y otro tipo de tumores muy vascularizados, como el rabdomiosarcoma o fibrosarcoma. Por tratarse de un embarazo de alto riesgo, se decide cesárea electiva a la semana 36. Nace un varón que no cursó con distrés respiratorio y que a la exploración física presentaba una gran tumoración alrededor del cuello y en la parte superior de la espalda. Era una lesión caliente no pulsátil (Fig. 8).
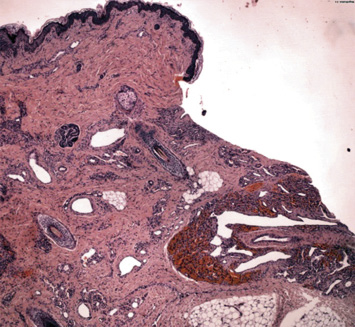
Figura 8. Exploraciones complementarias El ecocardiograma no mostró signos de insuficiencia cardíaca, la vena yugular se identificaba más ingurgitada y la carótida izquierda de mayor tamaño. La ecografía transfontanelar y abdominal no mostraron alteraciones. La analítica complementaria incluyendo plaquetas, hormonas tiroideas y dímero D fueron normales. La biopsia de piel mostró una proliferación vascular que se extendía hasta hipodermis, compuesta por vasos de diferente calibre. En profundidad, se observaba una zona con signos de hemorragia y un artefacto con aplastamiento de los vasos alargados en forma de asta de reno. No había grandes lóbulos celulares (Fig. 9).
Figura 9. La inmunohistoquímica mostró positividad para CD31 y D2-40 y el marcador tumoral GLUT-1 fue negativo. Resultados Los hallazgos clínicos e histopatológicos apoyan el diagnóstico de un hemangioendotelioma kaposiforme. Este es un interesante caso en el cual, la sospecha se contempló desde el periodo neonatal. Si bien, la clínica era sugestiva, fue necesaria la correlación clínico-patológica. Ante el gran tamaño y la posibilidad de presentar complicaciones como: insuficiencia cardíaca de alto gasto, obstrucción de la vía aérea o un fenómeno de Kasabach-Merritt, se decidió de manera multidisciplinaria, iniciar tratamiento con rapamicina sistémica con excelente respuesta clínica y poca toxicidad.
|
| Caso clínico 2 |
|
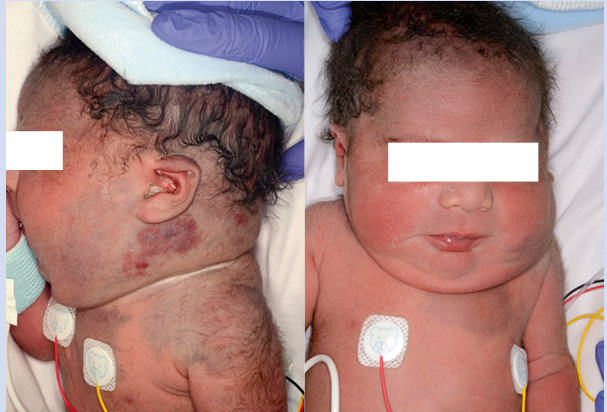
Lactante varón que a las 17 horas de vida inicia con distrés respiratorio y rechazo de tomas, que precisa ingreso para estudios complementarios. La radiografía de tórax inicial mostró velamiento de ambos campos pulmonares y cardiomegalia. El ecocardiograma evidenció hipertensión pulmonar (HTP) sin cardiopatía estructural. Se inició tratamiento con sildenafilo y, ante el empeoramiento clínico a las 62 horas de vida, requiere: intubación, ventilación mecánica y soporte inotrópico. A la semana, no había un diagnóstico claro de la causa de la HTP que indicara su deterioro progresivo, por lo que se decide descartar la presencia de una malformación arteriovenosa (MAV) a nivel sistémico. La ecografía transfontanelar mostró una imagen sugestiva de malformación vascular tipo fístula arteriovenosa dural o parenquimatosa. Antecedentes personales Perinatales: producto de primer embarazo a término (41 semanas), bien controlado. Serologías no reactivas y ecografías prenatales normales. Antecedentes familiares La madre, una tía materna, el abuelo materno y una prima de la madre presentaban máculas de aspecto vascular. Ningún familiar tenía estudios previos de resonancia magnética cerebral y/o espinal. Exploración física En piel, presentaba múltiples máculas de color rosa-parduzcas pálidas, de forma redondeada con un halo blanquecino periférico y de tamaños entre 1-2 cm, distribuidas en tronco y raíz de extremidades. Algunas estaban ligeramente calientes al tacto (Fig. 10).
Figura 10. Exploraciones complementarias La resonancia cerebral contrastada confirma la presencia de una fístula pial de alto flujo a nivel parietal izquierdo, con drenaje venoso hacia el seno sagital superior y un aporte arterial de una rama de la arteria cerebral media izquierda y también de una rama de la arteria cerebelosa anteroinferior (Fig. 11).
Figura 11. Se realizó embolización de las dos aferencias principales, lográndose una marcada disminución de flujo hacia la fístula. Diez días después, presenta nuevamente deterioro clínico con neuroimágenes que evidenciaron múltiples sangrados intracraneales, como hemorragia subaracnoidea e intraparenquimatosos, con algunas áreas de restricción de la difusión, sugestivos de lesión isquémica concomitante. Por todo ello, se procedió a la resección abierta de la fístula. Con la sospecha de fístula arteriovenosa como parte del síndrome de malformación capilar-malformación arteriovenosa (MC-MAV), se cursó estudio genético en sangre periférica y tejido resecado. Resultados Se confirmó mutación en heterocigosis de RASA1 (p.Arg903*/c.2707C>T) en línea germinal y en el tejido resecado. Las mutaciones en RASA1 se asocian en un 18% a la presencia de MAV o fístulas AV intracraneales y/o espinales (7,1 y 11,4%, respectivamente). A su vez, el 43% de los niños que cursan con hemorragias intracraneales secundarias a MAV tienen el antecedente familiar en primer grado de lesiones típicas de MC-MAV. La presencia de las lesiones cutáneas y la historia familiar de lesiones familiares hubiese podido sugerir el diagnóstico de este síndrome en el primer momento. En conclusión, se trata de un paciente que debutó con clínica secundaria a la fístula AV cerebral. En pacientes asintomáticos, el reconocimiento de las lesiones cutáneas obliga a la realización de pruebas de imagen para descartar MAV craneales y espinales antes de que den clínica. Así mismo, es importante realizar un examen de piel a todos los familiares. La penetrancia de esta mutación es prácticamente completa, por lo que una exploración cutánea normal, probablemente lo descartaría.
|





 Atopic dermatitis and seborrheic dermatitis
Atopic dermatitis and seborrheic dermatitis