 |
| Temas de FC |
A. Rodríguez Alonso, C. Molina Amores, M. Ruiz de Valbuena Maiz
Facultativos Especialistas de Área. Unidad de Patología Compleja. Servicio de Pediatría Hospitalaria, Enfermedades Infecciosas y Tropicales. Hospital Universitario La Paz, Madrid
| Resumen
Las patologías pulmonares crónicas conforman un grupo heterogéneo de enfermedades, entre las que se encuentran: neumopatías intersticiales, bronquiectasias no fibrosis quística, discinesia ciliar primaria y aspiración pulmonar crónica. Se trata de un grupo de enfermedades poco frecuentes, con manifestaciones clínicas inespecíficas y expresividad clínica variable, por lo que es importante mantener un alto índice de sospecha que permita reconocer los signos y síntomas de alarma. El diagnóstico suele ser difícil de establecer. No se dispone de tratamiento curativo y suelen estar asociadas a una alta morbimortalidad. Algunos casos pueden evolucionar hacia una neumopatía terminal. En estos casos, podría estar indicado el trasplante pulmonar. |
| Abstract
Chronic pulmonary pathologies make up a heterogeneous group of diseases, among which are interstitial lung diseases, non-cystic fibrosis bronchiectasis, primary ciliary dyskinesia and chronic pulmonary aspiration. It is a group of infrequent diseases, with nonspecific clinical manifestations and variable clinical expression, so it is important to maintain a high index of suspicion that allows the recognition of alarm signs and symptoms. The diagnosis is often difficult to establish. Curative treatment is not available and they are usually associated with high morbidity and mortality. Some cases can progress to end-stage lung disease. In these cases, lung transplantation may be indicated. |
Palabras clave: Neumopatías intersticiales; Bronquiectasias; Discinesia ciliar primaria; Aspiración respiratoria.
Key words: Interstitial lung diseases; Bronchiectasis; Respiratory aspiration; Ciliary motility disorders; Respiratory aspiration.
Pediatr Integral 2021; XXV (2): 101 – 108
Patología pulmonar crónica
Introducción
Las patologías pulmonares crónicas constituyen un grupo heterogéneo de enfermedades, generalmente poco frecuentes y con sintomatología inespecífica. A continuación, haremos una breve descripción de algunas de las más frecuentes o relevantes, excluyendo el asma y la fibrosis quística.
Neumopatías intersticiales
Introducción
Las neumopatías intersticiales en niños son enfermedades raras, crónicas y asociadas a elevada morbilidad y mortalidad. Se diagnostican con más frecuencia en el primer año de vida.
Las enfermedades pulmonares intersticiales son un grupo de enfermedades raras que afectan a los alvéolos y a los tejidos perialveolares, originando alteraciones del intercambio gaseoso. Constituyen un grupo numeroso de enfermedades, con más de 200 entidades. Se estima que la prevalencia es inferior a 1/100.000, en contraste con la prevalencia de 60-80/100.000 en adultos(1).
Su etiología es muy variada, incluyendo formas de origen genético, infeccioso, relacionadas con factores ambientales, fármacos, enfermedades sistémicas o de causa desconocida(2).
Aunque se pueden presentar en todas las edades, la edad de inicio más frecuente es el primer año de vida.
Clasificación
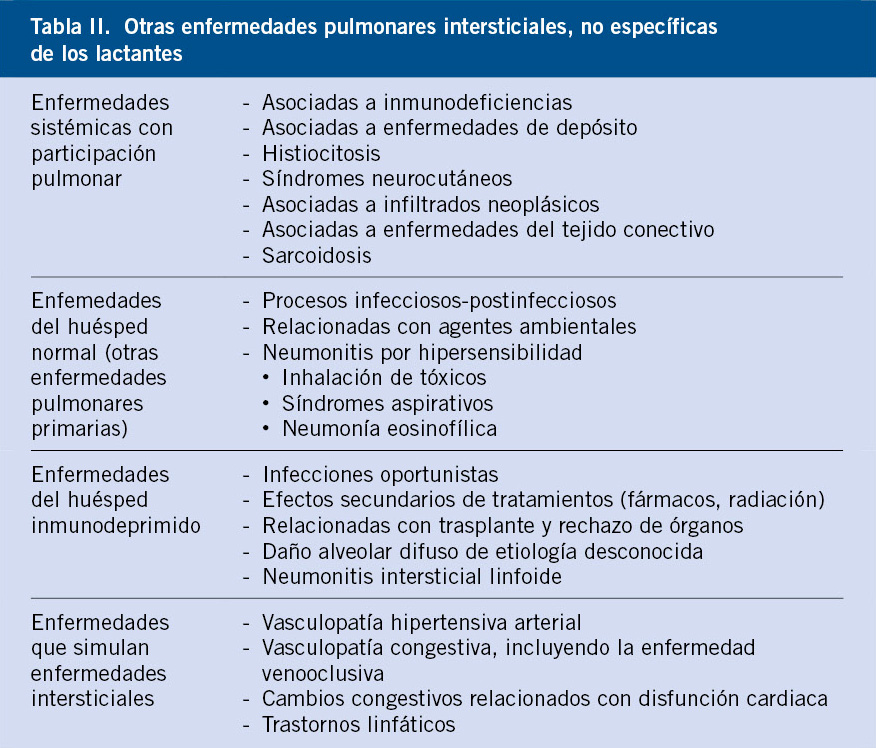
El consorcio americano chILD (Children’s Interstitial Lung Disease) ha propuesto una clasificación(3) en la que se incluyen: formas propias de los niños, enfermedades sistémicas con afectación pulmonar, enfermedades pulmonares primarias que afectan al huésped normal, enfermedades que afectan al huésped inmunodeprimido y enfermedades que simulan enfermedades intersticiales (afectación vascular pulmonar) (Tablas I y II).

Manifestaciones clínicas
El inicio suele ser sutil. La clínica se caracteriza por dificultad respiratoria con taquipnea, tiraje e hipoxemia.
La clínica suele ser insidiosa y poco específica, aunque, a veces, sobre todo en neonatos, se puede presentar de forma rápidamente progresiva.
En el recién nacido, se manifiesta como un cuadro de distrés respiratorio agudo con patrón alvéolo-intersticial bilateral en la radiografía de tórax, que a menudo requiere ventilación mecánica y que es indistinguible de una neumonía neonatal o de una enfermedad de membrana hialina(1).
En los primeros dos años de vida, la clínica se caracteriza por: taquipnea moderada, retraso ponderal, rechazo del alimento, tos seca, sibilantes en ausencia de infecciones respiratorias e hipoxemia nocturna y/o diurna(1).
Los niños de mayor edad pueden debutar con disnea o cianosis con el ejercicio, que progresa hasta aparecer en reposo.
A veces, los pacientes están relativamente asintomáticos y el diagnóstico se realiza, de forma casual, al realizar una radiografía de tórax por otros motivos.
En la auscultación, es habitual detectar estertores subcrepitantes y, ocasionalmente, algún sibilante, aunque puede ser normal en más de un tercio de los casos. Otros signos físicos incluyen: acropaquias y deformidad torácica, particularmente pectus excavatum, especialmente en las mutaciones del gen ABCA-3(4) (gen ATP binding cassette transporter A3).
Para facilitar el diagnóstico, el consorcio americano ha propuesto definirlo como: un síndrome (síndrome chILD) en el que son necesarias tres condiciones de entre las cuatro siguientes(3):
1. Síntomas respiratorios: tos, dificultad respiratoria y mala tolerancia al ejercicio.
2. Signos clínicos: taquipnea en reposo, estertores a la auscultación, tiraje, acropaquias, retraso en el crecimiento o insuficiencia respiratoria.
3. Hipoxemia con el ejercicio y/o en reposo.
4. Infiltrados difusos en la radiografía (Rx) o la tomografía computarizada (TC) de tórax.
El síndrome chILD requiere que se hayan descartado otras causas más frecuentes de enfermedad pulmonar difusa, como: fibrosis quística, inmunodeficiencias, cardiopatías congénitas, displasia broncopulmonar, infección pulmonar, discinesia ciliar primaria y aspiración recurrente(4).
Estudios diagnósticos
La TC pulmonar es la prueba más sensible para el estudio de las neumopatías intersticiales. El diagnóstico definitivo, a veces, será por biopsia pulmonar.
El diagnóstico se basa en las pruebas de imagen junto con: historia clínica, estudios de laboratorio y, a veces, lavado broncoalveolar y/o biopsia pulmonar (Fig. 1)(1).

Figura 1. Enfoque diagnóstico del niño con enfermedad pulmonar intersticial(1).
TC: Tomografia computarizada;
chILD: Children’s Interstitial Lung Disease; LBA: lavado broncoalveolar
• Rx de tórax. El 90% de los pacientes con enfermedad intersticial presentan alteraciones. En algunos casos, puede ser normal o mostrar alteraciones mínimas.
• La TC pulmonar es más sensible que la Rx de tórax. En algunos casos, permite el diagnóstico de la enfermedad, pero más a menudo, orienta sobre la causa. Es útil para valorar la gravedad y extensión de las lesiones y permite identificar el lugar idóneo a biopsiar. Además, es útil para controlar la respuesta terapéutica. Se recomienda realizar siempre TCAR (TC de alta resolución)(5).
• Pruebas de función pulmonar. No proporcionan datos para el diagnóstico etiológico, pero sí categorizan mejor la enfermedad y ayudan en el seguimiento. En general, suelen presentar un patrón restrictivo. En los lactantes con hiperplasia de células neuroendocrinas se ha descrito un patrón obstructivo con hiperinsuflación(5).
• Ecocardiografía. Debe ser una de las investigaciones iniciales a realizar para descartar hipertensión pulmonar y excluir anomalías vasculares y estructurales.
• Estudios analíticos. Incluye la realización de: estudio inmunológico, hipersensibilidad, autoinmunidad y enfermedades sistémicas.
• Broncoscopia flexible y lavado broncoalveolar. Son de especial utilidad para descartar infecciones en inmunodeprimidos y en algunas enfermedades(2) como: hemosiderosis, histiocitosis, neumonía eosinofílica, neumonitis por hipersensibilidad, sarcoidosis y proteinosis alveolar.
• Biopsia pulmonar quirúrgica(1,4). El diagnóstico definitivo y específico de las enfermedades intersticiales requiere, en muchos casos, del análisis histológico del parénquima pulmonar. La biopsia pulmonar quirúrgica está indicada cuando no se ha obtenido un diagnóstico específico con las exploraciones previas. Su indicación debe valorarse en cada caso en particular. Deben tomarse muestras de, al menos, dos áreas diferentes.
• Estudios genéticos(4). No se realizará de rutina, sino de forma selectiva según la sospecha clínica. Se han descrito mutaciones relacionadas con: déficit de proteínas del surfactante (déficit de proteína B o C y ABCA-3), mutaciones del gen NKX2.1 o TTF-1 asociadas con neumopatía intersticial, hipotiroidismo y alteración neurológica, mutaciones causantes de proteinosis alveolar, deficiencia de GATA2, mutaciones del gen filamin A (FLNA) y del gen ACTA2 que se asocian a pulmones con trastorno del desarrollo alveolar y mutaciones relacionadas con trastornos del desarrollo pulmonar: gen FOXF1.
Tratamiento
El tratamiento más utilizado son los corticoides.
Las medidas generales incluyen: corregir la hipoxemia, optimizar la nutrición, tratar las infecciones, promover la correcta inmunización (fundamentalmente, frente a gripe y neumococo) y evitar la exposición al tabaco(6).
El tratamiento más utilizado son los corticoides sistémicos. Se pueden usar pulsos de metilprednisolona de 10-30 mg/kg/día intravenosos durante 3 días consecutivos, mensualmente, durante 1 a 6 meses o prednisolona oral a dosis de 1-2 mg/kg/día, como tratamiento alternativo(5).
La hidroxicloroquina se ha mostrado también útil en algunos niños y se ha utilizado sola o en combinación con los corticoides.
El trasplante pulmonar está reservado para los casos de mal pronóstico sin otra alternativa terapéutica.
Pronóstico
La morbilidad y mortalidad asociada con el síndrome chILD (y otros tipos de enfermedad pulmonar difusa) es incierta. Se ha reportado una morbilidad persistente de 50% y mortalidad general de 30%(6).
Son de mal pronóstico los trastornos del desarrollo y crecimiento pulmonar, los déficits de proteína B del surfactante y déficit de ABCA3 de presentación neonatal, casos asociados a inmunodeficiencia y con pobre respuesta a corticoides(6).
Presentan buen pronóstico, la hiperplasia de células neuroendocrinas o la glucogenosis intersticial pulmonar (sin trastorno importante del desarrollo alveolar asociado) y, en general, los casos con buena respuesta a corticoides(6).
Discinesia ciliar primaria
Introducción
La discinesia ciliar primaria es una enfermedad rara, de origen genético y herencia autosómica recesiva, que se caracteriza por el deterioro de la función ciliar debido a defectos en su ultraestructura y/o funcionamiento. Como consecuencia, se produce un mal aclaramiento de las secreciones que favorece el desarrollo de infecciones respiratorias y otitis de repetición(7).
Se estima que afecta a 1 de cada 10.000, aunque la prevalencia no está claramente establecida y se trata de una entidad infradiagnosticada(7).
Clínica
Las otitis de repetición, la rinitis desde el periodo neonatal y la tos húmeda/productiva crónica diaria, son las manifestaciones clínicas más frecuentes en los pacientes con discinesia ciliar primaria.
La sintomatología es heterogénea, inespecífica y varía con la edad (Tabla III)(7,8). Es característico, el inicio de los síntomas respiratorios a edades tempranas y su persistencia(7,8).

Diagnóstico
No hay una prueba gold standard para el diagnóstico. La ATS (American Thoracic Society) y la ERS (European Respiratory Society) han sugerido, en los últimos años, guías y algoritmos diagnósticos que se basan en la combinación de exámenes complementarios complejos que, en la mayoría de los casos, deben ser realizados en centros con experiencia(7).
Exploraciones complementarias
• Medición del óxido nítrico exhalado nasal: suelen presentar niveles muy bajos. Relativamente fácil de realizar (dificultad en lactantes y niños pequeños) y no invasivo. Tiene una excelente sensibilidad (90-100%) y una buena especificidad (75-97%). De forma aislada, no descarta ni confirma el diagnóstico(7).
• Análisis vídeo-microscópico de alta velocidad: consiste en la valoración de la movilidad ciliar mediante una muestra nasal. Puede haber alteraciones sutiles, por lo que un resultado normal no descarta el diagnóstico(7).
• Microscopía electrónica de transmisión: es el método de referencia para evaluar la ultraestructura ciliar. Es diagnóstico si se detectan anomalías características (especificidad >99%). Sin embargo, hasta en un 15-20% de los casos pueden no detectarse anomalías(7).
• Estudio genético: hasta la fecha se han descrito unos 30 genes implicados y, cada vez son más las mutaciones descritas; aunque, actualmente, se detecta la presencia de mutaciones en alrededor de un 65% de los casos(7).
Tratamiento
Las técnicas para mejorar el aclaramiento de las secreciones y el tratamiento precoz de las infecciones son los pilares del tratamiento(9). Se recomienda la vacunación frente a neumococo y gripe(9).
El uso de tubos de timpanostomía es controvertido, debido a las complicaciones descritas en estos pacientes, como: otorrea mucopurulenta recurrente, timpanoesclerosis y perforación permanente de la membrana timpánica(9).
Existe evidencia limitada sobre el tratamiento quirúrgico. El trasplante pulmonar puede ser una opción en pacientes con neumopatía terminal(9).
Bronquiectasias no fibrosis quística (FQ)
Introducción
Históricamente, el término bronquiectasia se utilizaba para referirse a bronquios con dilataciones anómalas e irreversibles, diagnosticados mediante broncografía(10).
En el momento actual, las bronquiectasias se definen como: episodios persistentes o recurrentes (>3 al año) de tos productiva crónica (> 4 semanas), algunas veces con crepitantes gruesos a la auscultación y acropaquias, asociados a ratio broncoarterial > 0,8 en TC torácico(11).
Su prevalencia, fuera de la fibrosis quística, es desconocida, con un rango variable entre los 0,2-735 casos/100.000 niños(11).
Fisiopatología
Las infecciones respiratorias, las inmunodeficiencias primarias, la aspiración pulmonar crónica, la discinesia ciliar primaria y las malformaciones de la vía aérea son las causas más frecuentes.
Se considera que la patogénesis de las bronquiectasias se fundamenta en el modelo del “ciclo vicioso” propuesto por Cole (infección>inflamación>daño pulmonar>infección)(11,12).
Hasta en el 60% de los casos, se identifica una causa subyacente. Las infecciones, las inmunodeficiencias primarias, la aspiración crónica/recurrente, la discinesia ciliar y las malformaciones de la vía aérea, son las causas más frecuentes(11). En la tabla IV, se recogen las patologías más frecuentes que predisponen al desarrollo de bronquiectasias no FQ(10,13).

Clínica
Debe sospecharse la presencia de bronquiectasias en cualquier niño con tos crónica productiva e infecciones respiratorias de repetición.
La tos crónica húmeda es el síntoma más frecuente y está presente en prácticamente todos los pacientes. En la exploración física, pueden auscultarse crepitantes gruesos y pueden verse acropaquias(11,12).
Las infecciones respiratorias recurrentes son frecuentes. Los microorganismos más comunes son: Haemophilus influenzae, Streptococcus pneumoniae, Moraxella catarrhalis y Staphylococcus aureus. Pseudomonas aeruginosa es infrecuente en niños con bronquiectasias no fibrosis quística y su presencia suele ser sugestiva de enfermedad avanzada(14).
Diagnóstico
La prueba de elección para el diagnóstico es la TCAR de tórax.
• Diagnóstico radiológico. La TCAR de tórax es la prueba diagnóstica de elección. El hallazgo más característico es el ratio broncoarterial > 0,8. Otros hallazgos pueden ser: engrosamiento de las paredes bronquiales, imagen “en raíl de tren”, bronquios dilatados que se extienden hasta la periferia, imágenes “en anillo de sello”, niveles hidroaéreos en la vía aérea y perfusión en mosaico con áreas de atrapamiento aéreo(12).
• Diagnóstico etiológico. La determinación de la causa de las bronquiectasias es fundamental. El estudio básico incluye: hemograma y determinación de inmunoglobulinas, test del sudor y/o estudio genético de fibrosis quística, cultivo de secreciones (incluyendo estudio de micobacterias) y serología de Aspergillus. Según la sospecha clínica se podría ampliar estudio con: Mantoux, despistaje de discinesia ciliar primaria, evaluación de disfagia y reflujo gastroesofágico, broncoscopia, ampliación de estudio inmunológico, VIH y niveles de alfa-1 antitripsina.
Se recomienda la realización de espirometría al diagnóstico y durante el seguimiento para evaluar la progresión e identificar exacerbaciones. En la mayoría de los casos, se observa un patrón obstructivo(15).
Tratamiento
Las dos medidas principales son la fisioterapia y el tratamiento precoz de las infecciones respiratorias.
Es importante tratar la causa, en caso de conocerse. En cuanto al manejo de las bronquiectasias, el tratamiento es fundamentalmente médico y se basa en el drenaje de secreciones y en las medidas de control y prevención de las infecciones respiratorias (Tabla V)(15,16).
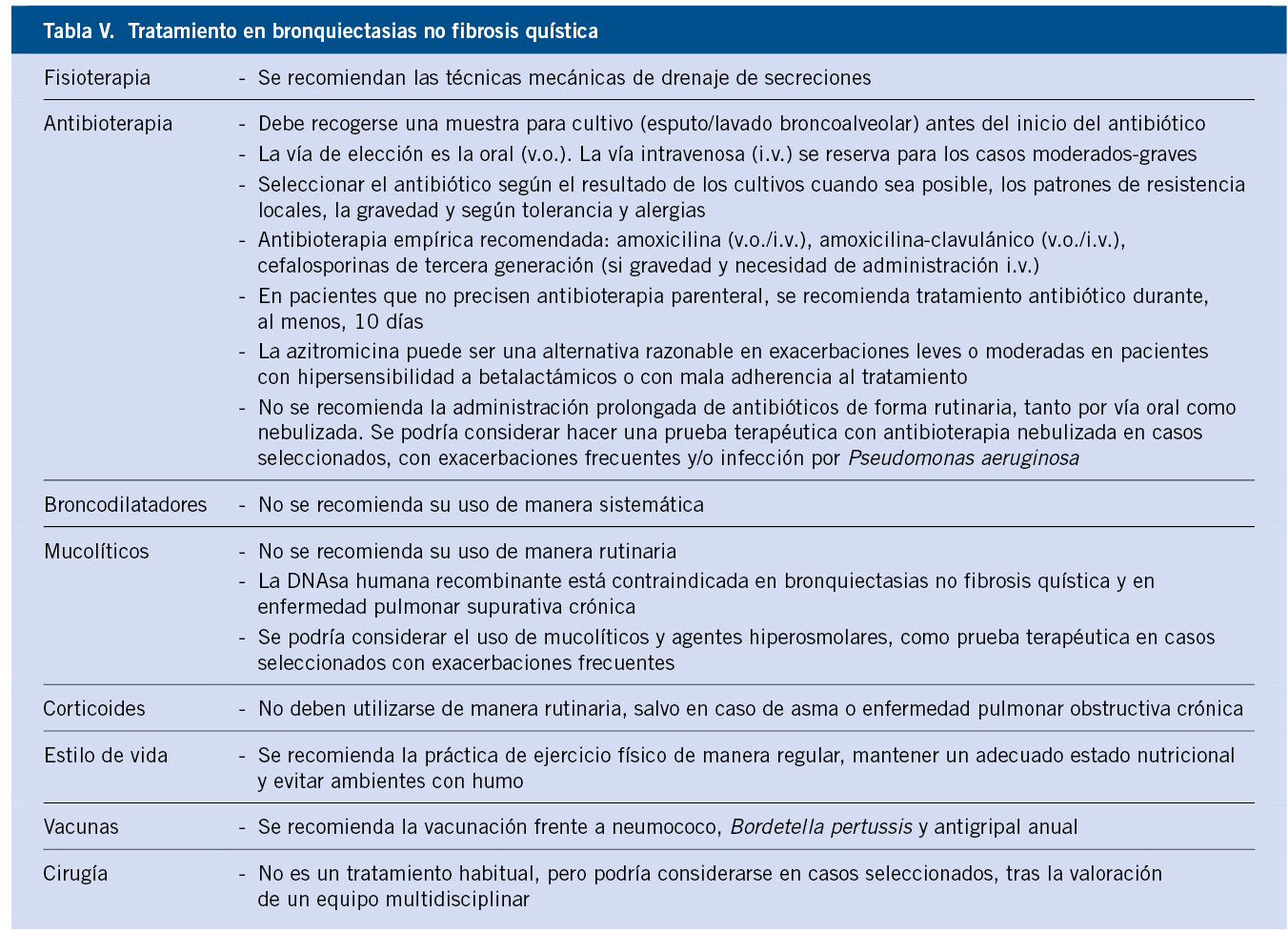
Las infecciones bacterianas juegan un papel importante en las exacerbaciones y progresión de las bronquiectasias, y constituyen un predictor independiente de deterioro de la función pulmonar, por lo que deben tratarse de manera precoz(15).
Pronóstico
Varios estudios en niños han demostrado que las bronquiectasias leves pueden ser reversibles, si se tratan de forma precoz e intensiva(16).
Neumopatía crónica aspirativa
Introducción
La aspiración pulmonar crónica consiste en el paso recurrente de material procedente de la cavidad oral (saliva, alimentos) o de reflujo gastroesofágico a la vía aérea inferior, que produce síntomas respiratorios crónicos o recurrentes(17-18).
Constituye una causa frecuente de morbilidad en niños con patología crónica y afectación multisistémica, y en niños con enfermedades neurológicas(18-19).
Fisiopatología
Se produce como consecuencia de enfermedad por reflujo y/o disfagia. Habitualmente, ocurre en pacientes con patologías subyacentes, especialmente en niños con afectación neurológica.
La aspiración pulmonar crónica ocurre en pacientes con enfermedad por: reflujo gastroesofágico, disfagia y/o mal manejo de secreciones oronasales, que tienen alterados los mecanismos protectores de la vía aérea(18).
La etiología suele ser multifactorial, con anomalías estructurales y/o funcionales que favorecen la aspiración (Tabla VI)(18-20).
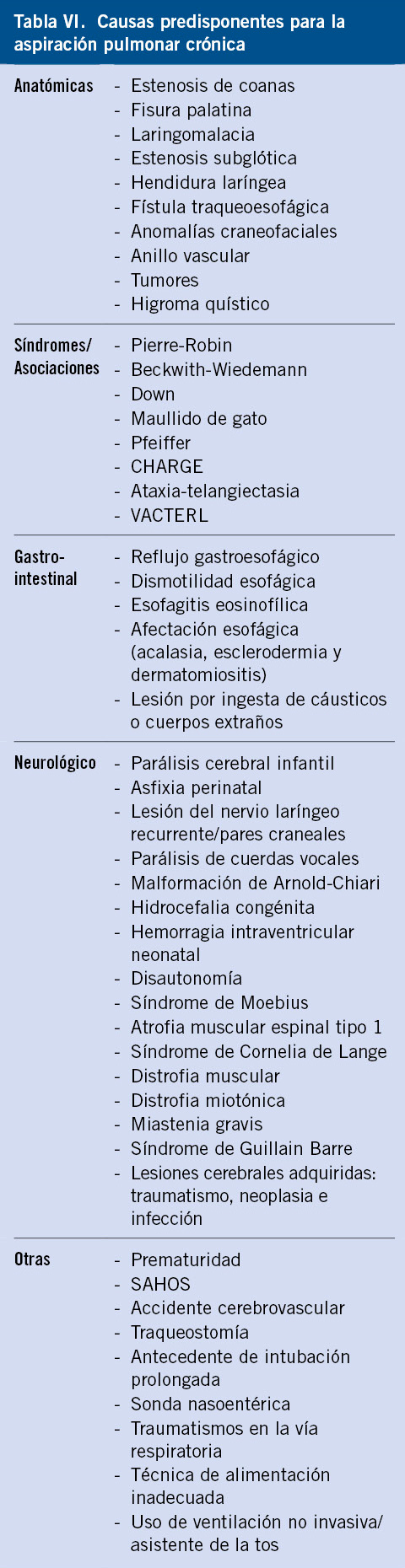
Clínica
Los síntomas más comunes son: tos crónica, estridor, disfonía, apneas o episodios de BRUE (Brief Resolved Unexplained Events), infecciones respiratorias de repetición, cuadros asmatiformes y fallo de medro. Son frecuentes los roncus y las sibilancias. En algunos casos, la aspiración es prácticamente silente(18-20).
La aspiración pulmonar crónica puede dar lugar a enfermedad pulmonar progresiva, bronquiectasias y fracaso respiratorio(20).
Diagnóstico
El diagnóstico es clínico, difícil de establecer, debido a la variabilidad de la sintomatología y a la ausencia de pruebas específicas. Se requiere un abordaje multidisciplinar y mantener un alto índice de sospecha.
El diagnóstico es clínico. Las pruebas complementarias deben seleccionarse según la sospecha clínica(18-19).
1. Historia clínica. Explorar, tanto síntomas pulmonares como extrapulmonares (deglución, reflujo gastroesofágico, sialorrea, desarrollo ponderoestatural), y detectar la presencia de factores predisponentes(18-19).
2. Pruebas complementarias:
• Evaluación del daño pulmonar: radiografía simple de tórax y TC de alta resolución. La TC es más sensible para la detección de lesiones en el parénquima. Típicamente, se ven afectados los segmentos apicales de los lóbulos inferiores y la parte posterior del lóbulo superior derecho. Los hallazgos que se pueden encontrar son: hiperinsuflación, engrosamiento bronquial, opacidades segmentarias, atelectasias, bronquiectasias, atrapamiento aéreo, consolidaciones, opacidades en vidrio deslustrado y engrosamiento pleural(18-19).
• Evaluación del mecanismo patogénico más probable:
- Reflujo gastroesofágico: pHmetría/impedanciometría, tránsito gastrointestinal superior y endoscopia digestiva superior.
- Trastorno deglutorio: evaluación clínica, fibroendoscopia de la deglución (FEES: fiberoptic endoscopic evaluation of sallowing) y videofluoroscopia.
- Aspiración de saliva: salivograma (poco sensible) y FEES(18-19).
• Fibrobroncoscopia flexible: de utilidad para la detección de anomalías anatómicas y la obtención de lavado broncoalveolar(18-19).
Tratamiento
Deberá estar orientado al tratamiento de la causa.
El tratamiento debe ser individualizado, dirigido al tratamiento de la causa y al mecanismo etiopatogénico(18-19).
• Reflujo gastroesofágico: medidas higiénico-dietéticas, tratamiento farmacológico (inhibidores de la bomba de protones, antagonistas de los receptores de histamina). La cirugía antirreflujo está indicada si ha fracasado el tratamiento farmacológico o en los casos de aspiraciones recurrentes o graves(18-19).
• Trastorno de la deglución: medidas posturales, reducción del volumen, uso de espesantes y rehabilitación. En caso de que con estas medidas la deglución no sea segura, estará indicado el uso de sondas de alimentación (nasogástrica, gastrostomía)(18-19).
• Aspiración de saliva: tratamiento farmacológico (glicopirrolato, parches de escopolamina, inyección de toxina botulínica en las glándulas salivales) o tratamiento quirúrgico (ligadura de conductos, extirpación de las glándulas salivales). En casos refractarios, se podría valorar la colocación de una cánula de traqueostomía con balón o, incluso, la cirugía de separación laringotraqueal(18-20).
Funciones del pediatra de Atención Primaria
• Identificación de pacientes con signos y síntomas compatibles con patología pulmonar crónica (tos crónica, infecciones respiratorias recurrentes, alteraciones en el patrón respiratorio fuera de procesos intercurrentes, anomalías radiológicas persistentes, etc.) y derivación a consultas de Neumología Pediátrica para estudio.
• Identificación y seguimiento evolutivo de los pacientes con factores de riesgo para el desarrollo de daño pulmonar crónico (pacientes con patología neuromuscular, síndromes polimalformativos, patología gastrointestinal, etc.) para su detección precoz.
• Reforzar la importancia de la fisioterapia respiratoria y garantizar el seguimiento en consultas de Rehabilitación.
• Tratamiento precoz y agresivo de las infecciones respiratorias intercurrentes.
• Asegurar una adecuada inmunización frente a gripe (vacunación del paciente y convivientes) y neumococo, inmunoprofilaxis frente a VRS en los casos en los que se considere indicado, y recomendar evitar la exposición al humo del tabaco y otros irritantes de la vía aérea.
Bibliografía
Los asteriscos muestran el interés del artículo a juicio de las autoras.
1.*** Bush A, Cunningham S, de Blic J, Barbato A, Clement A, Epaud R, et al. European protocols for the diagnosis and initial treatment of interstitial lung disease in children. Thorax. 2015; 70: 1078-84.
2.** Liñán Cortés S, Moreno Galdó A. Neumopatías intersticiales. En: Andrés Martín A, Valverde Molina J. Manual de Neumología Pediátrica. Madrid: Editorial Panamericana S.A.; 2011. p.361-73.
3.* Kurland G, Deterding RR, Hagood JS, Young LR, Brody AS, Castile RG, et al. An Official American Thoracic Society Clinical practice guideline: classification, evaluation, and management of childhood interstitial lung disease in infancy. Am J Respir Crit Care Med. 2013; 188: 376-94.
4.*** Moreno Galdó A, de Mir Messa I, Liñán Cortes S. Neumopatía intersticial. Sospecha clínica y abordaje. Protoc Diagn Ter Pediatr. 2017; 1: 221-35.
5. Comité Nacional de Neumonología. Enfermedades del intersticio pulmonar en niños menores de 2 años. Clasificación, diagnóstico y tratamiento. Arch Argent Pediatr. 2019; 117: S120-S134.
6. Rubilar L, Maggiolo J. Enfermedad pulmonar intersticial del lactante. Neumol Pediatr. 2014; 9: 21-6.
7.*** Kuehni CE, Lucas JS. Diagnosis of primary ciliary dyskinesia: summary of the ERS Task Force REPORT. Breathe. 2017; 13: 166-78.
8.** Fitzgerald DA, Shapiro AJ. When to suspect primary ciliary dyskinesia in children. Pediatric Respiratory Reviews. 2016; 18: 3-7.
9.** Lucas JS, Alanin MC, Collins S, Harris A, Johansen HK, Nielsen KG, et al. Clinical care of children with primary ciliary dyskinesia. Expert Review of Respiratory Medicine. 2017; 11: 779-90.
10.* Bush A, Floto RA. Pathophysiology, causes and genetics of paediatric and adult bronchiectasis. Respirology. 2019; 24: 1053-62.
11.* Poeta M, Maglione M, Borrelli M, Santamaria F. Non-cystic fibrosis bronchiectasis in children and adolescents: Neglected and emerging issues. Pediatrics & Neonatology. 2020; 61, 255-62.
12.*** Chang AB, Bush A, Grimwood K. Bronchiectasis in children: diagnosis and treatment. Lancet. 2018; 392: 866-79.
13. Brower KS, Del Vecchio MT, Aronoff SC. The etiologies of non-CF bronchiectasis in childhood: a systematic review of 989 subjects. BMC Pediatr. 2014; 14: 4.
14. Kapur N, Grimwood K, Masters IB, Morris PS, Chang AB. Lower airway microbiology and cellularity in children with newly diagnosed non-CF bronchiectasis. Pediatr Pulmonol. 2012; 47: 300-7.
15.*** Chang AB, Bell SC, Torzillo PJ, King PT, Maguire GP, Byrnes CA, et al. Chronic suppurative lung disease and bronchiectasis in children and adults in Australia and New Zealand Thoracic Society of Australia and New Zealand guidelines. Med J Aust. 2015; 202: 21-3.
16.** Wu J, Chang AB, Wurzel DF. Contemporary management of bronchiectasis in children. Expert Rev Respir Med. 2019; 13: 969-79.
17.*** Torres-Silva CA. Chronic pulmonary aspiration in children: diagnosis and management. Curr Probl Pediatr Adolesc Health Care. 2018; 48: 74-81.
18.*** Huidobro B, Huerta J, Barredo E, Tolín M, Rodríguez-Cimadevilla J, Salcedo A. Manejo del síndrome aspirativo recurrente en el paciente pediátrico. Acta Pediatr Esp. 2009; 67: 420-6.
19.*** Cuestas G, Rodríguez V, Bellia Munzón P, Bellia Munzón G. Algoritmo para el manejo de la aspiración pulmonar crónica en pediatría. Arch Argent Pediatr. 2019; 117: 412-20.
20.*** Tutor JD. Dysphagia and chronic pulmonary aspiration in children. Pediatrics in Review. 2020; 41: 236-44.
Bibliografía recomendada
- Bush A, Cunningham S, de Blic J, Barbato A, Clement A, Epaud R, et al. European protocols for the diagnosis and initial treatment of interstitial lung disease in children. Thorax. 2015; 70: 1078-84.
Propuesta de procedimiento diagnóstico en el niño con sospecha de neumopatía intersticial, con la colaboración de chILD-EU y protocolo de tratamiento, siguiendo el método Delphi (consenso tras encuestas a clínicos de diferentes países).
- Moreno Galdó A, de Mir Messa I, Liñán Cortes S. Neumopatía intersticial. Sospecha clínica y abordaje. Protoc Diagn Ter Pediatr. 2017; 1: 221-35.
Enfoque diagnóstico según la edad de presentación y revisa las diferentes entidades clínicas específicas de las neumopatías intersticiales.
- Kuehni CE, Lucas JS. Diagnosis of primary ciliary dyskinesia: summary of the ERS Task Force REPORT. Breathe. 2017; 13: 166-78.
Sintetiza, de una forma muy completa y sencilla, la discinesia ciliar primaria y, posteriormente, resume las recomendaciones de la Task Force sobre el diagnóstico de la misma.
- Chang AB, Bush A, Grimwood K. Bronchiectasis in children: diagnosis and treatment. Lancet. 2018; 392: 866-79.
Artículo muy completo y didáctico sobre las bronquiectasias, en el que se describe, desde la epidemiología hasta el seguimiento. Cuenta con varias tablas y gráficos que hacen que sea un artículo muy visual, entretenido y fácil de leer.
- Chang AB, Bell SC, Torzillo PJ, King PT, Maguire GP, Byrnes CA, et al. Chronic suppurative lung disease and bronchiectasis in children and adults in Australia and New Zealand Thoracic Society of Australia and New Zealand guidelines. Med J Aust. 2015; 202: 21-3.
Guías clínicas más recientes para el manejo de la enfermedad pulmonar supurativa crónica y bronquiectasias no fibrosis quística, tanto en adultos como en niños. Cuenta con definiciones, recomendaciones acerca de cuándo derivar a un paciente para despistaje de: bronquiectasias, pruebas complementarias, tratamiento y otras recomendaciones.
- Cuestas G, Rodríguez V, Bellia Munzón P, Bellia Munzón G. Algoritmo para el manejo de la aspiración pulmonar crónica en pediatría. Arch Argent Pediatr. 2019; 117: 412-20.
Artículo de revisión muy completo. Cuenta con tablas en las que se resumen las causas de disfagia y las diferentes pruebas complementarias en la aspiración crónica y algoritmos para el manejo de la enfermedad.
| Caso clínico |
|
Adolescente de 13 años que acude a consulta para valoración por infecciones respiratorias de repetición. Antecedentes personales • Parálisis cerebral infantil tipo tetraparesia espástica. Nivel V de la GMFSC (Gross Motor Function Classification System). • Epilepsia. • Encefalopatía hipóxico-isquémica. • Desnutrición crónica grave. • Tratamiento con budesonida nebulizada 400 mcg/12 h. • Vacunación según calendario + neumococo. Vacunación antigripal estacional y convivientes. • No alergias. Anamnesis por aparatos • Respiratorio: cuatro ingresos por neumonías con necesidad de antibioterapia intravenosa y oxigenoterapia suplementaria. Última hospitalización con ingreso en UCIP y necesidad de ventilación no invasiva. Múltiples ciclos de tratamiento antibiótico, broncodilatador y corticoide oral, por cuadros de: tos húmeda recurrente, fiebre/febrícula y sibilancias. Su pediatra inició tratamiento con budesonida inhalada 400 mcg/12 h sin mejoría. Tos húmeda crónica diaria. No ronquido ni apneas. • Digestivo: come por boca dieta tamizada. En general, tose con las comidas, tarda más de hora y media en comer. No vómitos frecuentes, pero sí rumiación. Llanto tras las comidas y posturas distónicas. Sialorrea. Hábito estreñido, usan enemas cada 4-5 días, si no hace deposición. Heces de tipo 1 en la escala de Bristol.. Exploración física SatO2: 93%. FR: 25 rpm. Peso: 17 kg. Buen estado general. Desnutrida. Cifoescoliosis izquierda. Bien hidratada y perfundida. ACP: ruidos cardiacos rítmicos sin soplos. Aceptable ventilación bilateral. Roncus dispersos. Orofaringe y otoscopia normales. Abdomen normal. Neurológico: Tetraparesia espástica. No tiene sostén cefálico. Pie equinovaro bilateral. Deformidad en carpos. Exploraciones complementarias • Radiografía de tórax: engrosamientos peribronquiales y marcado aumento de la trama broncovascular en ambas bases. • TC pulmonar: bronquiectasias múltiples de aspecto varicoso / quístico que ocupan difusamente el LID. Igualmente, existen bronquiectasias en segmento posterior del LSD y, en menor grado, en el resto de los segmentos a nivel central. El pulmón izquierdo tiene bronquiectasias en menor número que en el lado contralateral, aunque también son significativas, de aspecto cilíndrico y varicoso con predominio en LII. Atrapamiento aéreo con hiperventilación de vertiente posterior del LSI y LII.
|

 New approach in the treatment of children with asthma
New approach in the treatment of children with asthma