 |
| Temas de FC |
A. López Neyra*, A. Lamas Ferreiro**
*Médico Adjunto. Unidad de Fibrosis Quística. Sección de Neumología Pediátrica. Hospital Infantil Universitario Niño Jesús. Madrid. **Médico Adjunto. Unidad de Fibrosis Quística. Neumología Pediátrica. Instituto Ramón y Cajal de Investigación Sanitaria (IRYCIS). Hospital Universitario Ramón y Cajal. Madrid
| Resumen
La fibrosis quística (FQ) es una enfermedad genética causada por mutaciones en el gen CFTR. La alteración de la proteína CFTR modifica el transporte iónico a nivel epitelial, dando lugar a secreciones espesas en los órganos donde se expresa: pulmón, tubo digestivo, hígado o glándulas sudoríparas, entre otros. En la vía aérea, estas secreciones espesas condicionan la tríada de: obstrucción, infección e inflamación crónicas, responsable de la destrucción tisular, fallo respiratorio y, finalmente, muerte del paciente. La bacteria más frecuente en la infección bronquial crónica es Pseudomonas aeruginosa. Además de la infección crónica, pueden existir otras complicaciones pulmonares, como: neumotórax, atelectasia, hemoptisis o aspergilosis broncopulmonar alérgica, entre otras. |
| Abstract
Cystic fibrosis (CF) is a genetic disease caused by mutations in the CFTR gene. The abnormal CFTR protein modifies ion transportation at the epithelial level, giving rise to thick secretions in the organs where it is expressed: lungs, digestive tract, liver and sweat glands, among others. In the airway, these thick secretions condition the triad of obstruction, infection, and chronic inflammation, responsible for tissue destruction, respiratory failure and, lastly, the patient ́s death. The bacteria most frequently found in chronic bronchial infections is Pseudomonas aeruginosa. In addition to chronic infection, there may be other pulmonary complications such as pneumothorax, atelectasis, hemoptysis, or allergic bronchopulmonary aspergillosis, among others. |
Palabras clave: Fibrosis quística; CFTR; Infección; Complicaciones respiratorias; Pseudomonas aeruginosa.
Key words: Cystic fibrosis; CFTR; Infection; Respiratory complications; Pseudomonas aeruginosa.
Pediatr Integral 2021; XXV (2): 91 –100
Fibrosis quística y sus manifestaciones respiratorias
Introducción
La fibrosis quística es una enfermedad genética causada por mutaciones en el gen CFTR. La alteración de la proteína CFTR modifica el aclaramiento mucociliar, dando lugar a la aparición de secreciones espesas en los órganos donde se expresa.
La fibrosis quística (FQ) es una enfermedad autosómica recesiva, con una incidencia aproximada en nuestro medio de 1 de cada 5.000 recién nacidos vivos (datos no publicados) y una prevalencia de portadores heterocigotos aproximada en 1 de cada 40-50 personas en la raza caucásica(1,2). Está causada por mutaciones en el gen CFTR (regulador de la conductancia transmembrana de la FQ, del inglés Cystic Fibrosis Transmembrane conductance Regulator), localizado en el brazo largo del cromosoma 7, que codifica para una proteína del mismo nombre (CFTR) de 1.480 aminoácidos. Esta proteína se comporta como un canal de cloro e, indirectamente, controla los movimientos del sodio, bicarbonato y agua a través de la membrana celular(2). Hasta el momento, se han identificado más de 2.000 mutaciones diferentes agrupadas en 7 clases funcionales(3) (Tabla I), aunque no se conoce el significado patogénico de todas ellas.

Las clases I, II, III, VI y VII se suelen asociar con fenotipos graves de la enfermedad; las mutaciones de clases IV y V suelen tener cierta función residual y condicionar una enfermedad menos grave, asociada habitualmente a suficiencia pancreática(4). Existe una herramienta online (https://cftr2.org) en la que se pueden consultar las diferentes mutaciones y su significado patogénico. La mutación Phe.508del (clase II, antiguamente denominada F508del) es la más frecuente (>90% de los pacientes en todo el mundo presentan esta mutación en, al menos, 1 de los dos alelos). El resto de mutaciones son muy poco frecuentes y solo 5 de ellas tienen una prevalencia >1%(2). La prevalencia de Phe.508del no es homogénea y varía de unos países a otros. Según los datos del registro europeo, en España, el porcentaje de pacientes con, al menos, 1 alelo Phe.508del es de alrededor del 75%. Las siguientes mutaciones en función de su prevalencia en España son Gly542X (G542X) y Arg334Trp (R334W)(5).
La fisiopatología de la FQ está condicionada por el insuficiente o nulo funcionamiento de la proteína CFTR. La alteración del transporte de cloro determina una reabsorción elevada de sodio y agua, lo que da lugar a una secreción espesa, deshidratada y gruesa en la superficie de las células epiteliales donde se expresa: pulmón, tubo digestivo, hígado, glándulas sudoríparas, conductos deferentes o senos paranasales, entre otros(1,2).
Aunque las manifestaciones respiratorias de la FQ son las responsables de la mayoría de la morbimortalidad por esta enfermedad, el espectro de síntomas incluye: insuficiencia pancreática, enfermedad hepática y diabetes relacionada con la FQ, infertilidad masculina por agenesia de los conductos deferentes, obstrucción intestinal distal, íleo meconial o episodios de deshidratación por la pérdida excesiva de electrolitos por el sudor(1). El fenotipo final de cada paciente con FQ no solo está determinado por el defecto genético de CFTR, sino que se ve influido por otros factores, tanto ambientales como del propio enfermo. Estos factores explican que no exista siempre una buena correlación entre el genotipo y el fenotipo de los pacientes, sobre todo, en relación con la afectación respiratoria(1).
El desarrollo de las unidades multidisciplinares y la mejora del tratamiento nutricional, de la fisioterapia y del control de la infección bronquial, han determinado una disminución progresiva de la morbilidad de los pacientes con FQ en las últimas décadas, así como un aumento de su esperanza de vida, que actualmente se sitúa por encima de los 40 años(6).
Diagnóstico de la fibrosis quística
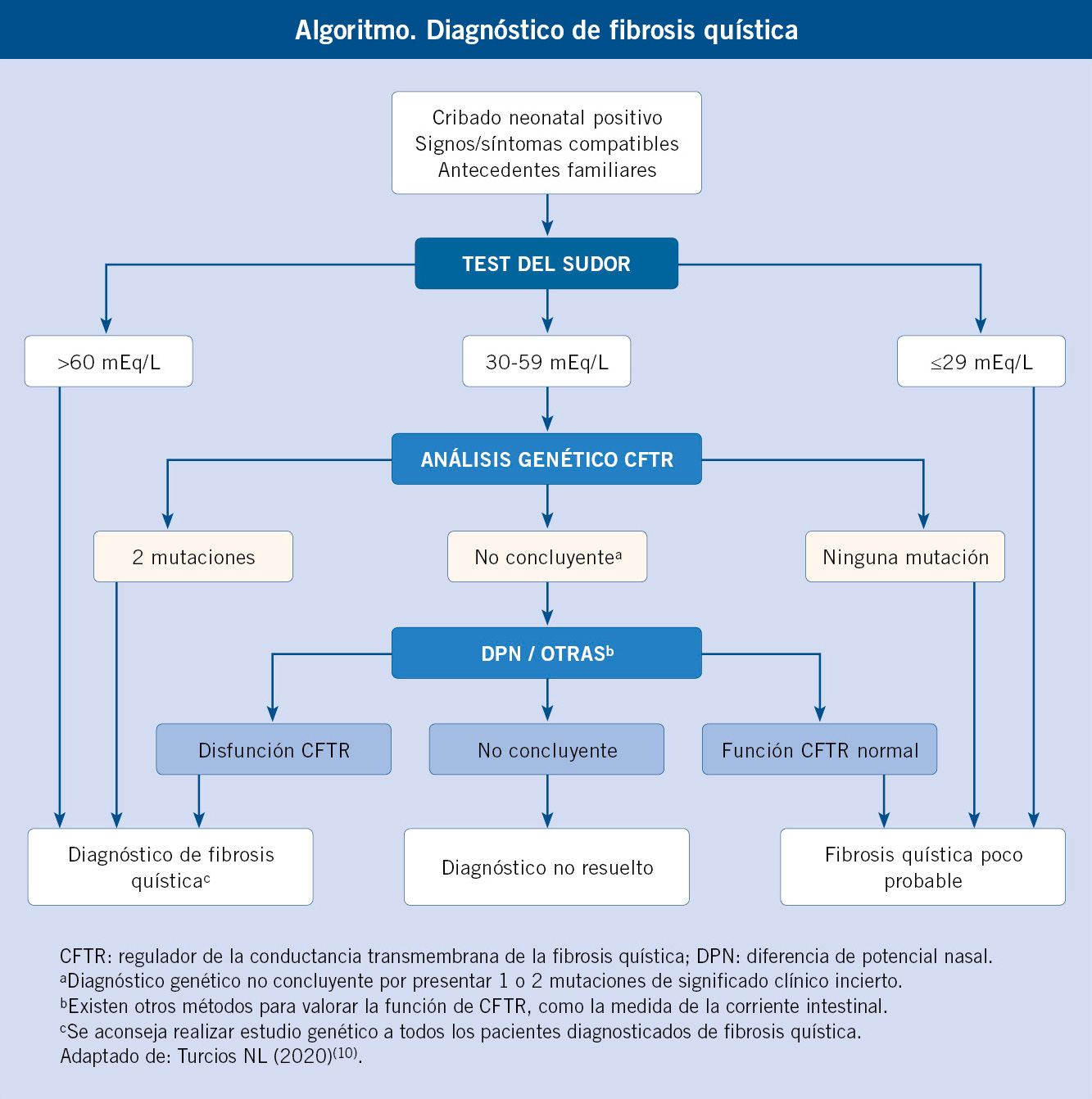
La mayoría de los diagnósticos actuales en fibrosis quística se realizan a través de los diferentes programas de cribado neonatal de cada Comunidad Autónoma. La concentración de cloro en sudor continúa siendo fundamental para el diagnóstico.
Para realizar el diagnóstico de FQ deben existir, en primer lugar, síntomas y/o signos compatibles con la enfermedad (Tabla II), antecedentes familiares de FQ en hermanos o primos, o un resultado positivo en una prueba de cribado neonatal.

A continuación, debe demostrarse la disfunción de la proteína CFTR mediante una prueba de laboratorio: concentración de cloro en sudor superior a 60 mEq/L, detección de dos mutaciones reconocidas de FQ o demostración de alteraciones en la diferencia de potencial nasal transepitelial(7) (Algoritmo 1).
En la actualidad, existen programas de cribado neonatal en todas las Comunidades Autónomas, por lo que la mayoría de los diagnósticos de FQ se realizan al nacer. El cribado neonatal es una herramienta fundamental para el diagnóstico temprano de la FQ, ofrece la oportunidad de iniciar el tratamiento de forma muy precoz e impacta de forma positiva en la evolución y el pronóstico de la enfermedad(8). El cribado neonatal se basa en la determinación de tripsinógeno inmunorreactivo en la sangre de talón y en la confirmación diagnóstica de FQ mediante la prueba del sudor con clorimetría. La introducción del análisis genético de CFTR mejora la sensibilidad y la especificidad de los programas de cribado, aunque aumenta la detección de portadores(4).
Fisiopatología de la enfermedad pulmonar
La alteración del aclaramiento mucociliar por el déficit o mal funcionamiento de CFTR, da lugar a secreciones espesas en el pulmón que favorecen la obstrucción, infección e inflamación, así como la destrucción tisular, el fallo respiratorio y la muerte en estos pacientes.
La enfermedad pulmonar en la FQ afecta inicialmente a las vías aéreas; los alveolos y el intersticio se ven afectados en las etapas más avanzadas de la enfermedad.
El aclaramiento mucociliar (AMC) normal requiere de la acción coordinada de los cilios de las células epiteliales bronquiales, que desplazan proximalmente la capa de moco que los recubre. Esta capa está formada por el moco, propiamente dicho, que descansa sobre una capa acuosa o líquido periciliar, en la que se mueven los cilios. La disfunción de CFTR y la alteración del transporte iónico normal producen una disminución de la excreción de cloro y un aumento de la absorción de sodio y agua, que en última instancia provocan: deshidratación del líquido periciliar, disminución de su altura y dificultad para el movimiento de los cilios. Este proceso impide un AMC y condiciona la obstrucción de la luz bronquial, lo que favorece la infección bacteriana crónica y la instauración de una respuesta inflamatoria exagerada y mal regulada, que también tiende a cronificarse. Este círculo de obstrucción-infección-inflamación pulmonar crónico provoca la progresiva destrucción del tejido pulmonar, la obstrucción crónica del flujo aéreo y la aparición de bronquiectasias, entre otras complicaciones, y finalmente, la insuficiencia respiratoria crónica y el fallecimiento del paciente. La disfunción de CFTR está relacionada no solo con la deshidratación de las secreciones, sino que también altera el pH de las mismas, lo que dificulta la inmunidad innata y tiene un papel directo en la instauración y el mantenimiento de la inflamación crónica(1,2). La inflamación en la FQ está dirigida fundamentalmente por los neutrófilos (aunque cada vez se conoce la implicación de otros tipos celulares, como los macrófagos o las células epiteliales) y se caracteriza por el desequilibrio entre factores proinflamatorios, como el factor de necrosis tumoral alfa (TNF-α), las interleucinas (IL) IL-8, IL-1β o IL-17, y factores antiinflamatorios como IL-10, lipoxinas o resolvinas(9).
Manifestaciones respiratorias de la fibrosis quística
La tos húmeda crónica es el síntoma más frecuente y característico de la fibrosis quística.
La afectación pulmonar es, sin duda, la más importante de la FQ, ya que condiciona la morbilidad y mortalidad por esta enfermedad. Aunque puede iniciarse en el primer año de vida, es a partir de la adolescencia cuando progresa de manera más evidente(10).
El síntoma respiratorio más frecuente en los pacientes con FQ es la tos, que característicamente es húmeda, asociada a secreciones respiratorias y de predominio matutino. En la infancia, la tos suele ser intermitente, asociada o desencadenada por infecciones respiratorias, y con buena respuesta a los tratamientos antibióticos orales. En el adolescente y adulto, la tos húmeda suele ser constante a lo largo del día y tiende a persistir, a pesar del tratamiento antibiótico. La tos se acompaña de expectoración, habitualmente espesa y de color variable, con un tono más amarillo-verdoso en situaciones de infección bronquial crónica. La expectoración se hace más abundante y persistente a medida que avanza la enfermedad. La disnea y la intolerancia al ejercicio se asocian con la afectación pulmonar grave, y suelen aparecer en fases avanzadas de la enfermedad.
Además de los síntomas respiratorios descritos, los pacientes con FQ presentan empeoramientos más o menos bruscos de su situación respiratoria, llamados exacerbaciones pulmonares (EP). Aunque no existe una definición consensuada de EP, los síntomas que deben hacer sospechar la existencia de una se resumen en la Tabla III.

El número y la intensidad de las EP influyen de manera importante en el pronóstico de la enfermedad(1,10), por lo que los tratamientos que retrasen o disminuyan las EP son fundamentales para el manejo de la FQ.
La infección bronquial crónica
Las condiciones de la vía aérea en los pacientes con fibrosis quística favorecen la infección bronquial crónica por bacterias, como Staphylococcus aureus o Pseudomonas aeruginosa.
La alteración del AMC favorece la infección bronquial crónica por distintas bacterias. El círculo de obstrucción-infección-inflamación genera un ambiente con baja tensión de oxígeno y un pH ácido que: favorece el crecimiento bacteriano, dificulta la acción del sistema inmune y condiciona la aparición de la infección bronquial crónica(11). En general, las especies aisladas en las muestras de pacientes con FQ, con el tiempo, presentan mayores resistencias que las aisladas en otras patologías.
La cronología bacteriana de la infección bronquial crónica en la FQ está bastante bien definida y sigue un patrón similar en la mayoría de los pacientes(12). En los primeros años, predominan las infecciones por Staphylococcus aureus (S. aureus) y Haemophilus influenzae, aunque pueden encontrarse otras bacterias, como Moraxella catarrhalis o Streptococcus pneumoniae. A partir de la adolescencia, aparecen las bacterias típicas de la FQ, entre las cuales, la más característica es Pseudomonas aeruginosa (P. aeruginosa)(10). La historia natural de la infección por P. aeruginosa comienza con una primoinfección, que habitualmente ocurre durante la infancia y cuya erradicación suele tener éxito. Con el tiempo, P. aeruginosa desarrolla mecanismos de adaptación y resistencia, los cultivos positivos se hacen más frecuentes y, finalmente, se establece la infección bronquial crónica, definida por la presencia de P. aeruginosa en más de la mitad de cultivos recogidos a lo largo de un año(13). Otras bacterias típicas de la FQ y que rara vez se encuentran en otras patologías respiratorias, son: Achromobacter xylosoxidans, Stenotrophomonas maltophilia o el complejo Burkholderia cepacia(12). Todas ellas son bacterias multirresistentes con escasas opciones terapéuticas, sobre todo, por vía oral. La infección por S. aureus resistente a la meticilina no es frecuente en nuestro medio, pero se ha descrito hasta en el 5% de los pacientes del registro europeo(5). Las micobacterias, sobre todo, Mycobacterium abscessus, son infecciones de difícil tratamiento con importantes implicaciones pronósticas para el paciente. La infección por hongos es frecuente, aunque su significación patológica no está del todo aclarada. La presencia de Aspergillus fumigatus en el esputo se ha asociado con infección aguda (bronquitis por Aspergillus) y, con más frecuencia, con el desarrollo de aspergilosis broncopulmonar alérgica (ABPA) secundaria, como reacción a la presencia de Aspergillus fumigatusen la vía aérea(14).
Exploración física y pruebas complementarias
La espirometría y el cultivo de secreciones respiratorias son fundamentales en el seguimiento del paciente con fibrosis quística.
Exploración física
La exploración física suele ser normal en los primeros años de la enfermedad. A partir de la adolescencia o cuando el paciente presenta tos y expectoración crónicas, pueden encontrarse crepitantes y/o sibilancias en la auscultación. La insuficiencia respiratoria crónica puede manifestarse por la aparición de alteraciones en la configuración del tórax, como el aumento de su diámetro anteroposterior secundario a la hiperinsuflación pulmonar o la aparición de escoliosis, cuya prevalencia es mayor que en la población general. En los pacientes con FQ pueden aparecer acropaquias a lo largo de la evolución.
Existen diferentes pruebas diagnósticas que deben realizarse tanto en el diagnóstico como en el seguimiento de las complicaciones respiratorias.
Función pulmonar
Los cambios en la espirometría forzada, sobre todo, del volumen espiratorio forzado en el primer segundo (FEV1) permiten valorar cambios agudos, hacer seguimiento a largo plazo y valorar la respuesta a los distintos tratamientos utilizados. Aunque, en los primeros años, la espirometría suele ser normal, con el tiempo se establece un patrón obstructivo progresivo que se relaciona bien con el número de las EP y con la mortalidad. El índice de aclaramiento pulmonar (LCI, del inglés lung clearance index), calculado a través del lavado alveolar de múltiples respiraciones, es una prueba más sensible que la espirometría en lactantes y prescolares(15).
Radiología
La radiografía de tórax es una prueba de imagen sencilla y accesible, pero poco sensible a la hora de valorar cambios. Aunque hace años se realizaba de forma sistemática cada 1-2 años, su uso para el seguimiento de la enfermedad pulmonar cada vez es menos frecuente. La radiografía de tórax sigue siendo muy útil para el diagnóstico de las complicaciones agudas, como la atelectasia o el neumotórax. La tomografía computarizada (TC) de tórax es mucho más sensible que la radiografía para detectar las alteraciones pulmonares y puede visualizarlas de forma más precoz. Los cortes en espiración permiten el diagnóstico de atrapamiento aéreo en edades muy tempranas. Tiene el inconveniente de que es una prueba menos accesible y con una mayor dosis de radiación, aunque los protocolos de baja intensidad con pocos cortes pueden disminuirla de forma significativa(16).
Microbiología
Los cultivos de esputo o de aspirado nasofaríngeo profundo en los pacientes más pequeños, deben realizarse de forma periódica, al menos, 4 veces al año(7). Son de utilidad para la detección precoz de bacterias que requieren un tratamiento específico, aun en ausencia de síntomas, así como para guiar los tratamientos antibióticos en caso de EP. Las muestras respiratorias de FQ se procesan de forma especial y, por tanto, es importante enviarlas a laboratorios con experiencia en FQ y con protocolos específicos(7).
Tratamiento de la enfermedad respiratoria
La hidratación de las secreciones respiratorias, la fisioterapia y el tratamiento antibiótico son los pilares fundamentales del tratamiento de la enfermedad respiratoria en la fibrosis quística.
El objetivo fundamental del tratamiento de la enfermedad respiratoria en la FQ, es retrasar la progresión de la enfermedad y evitar o disminuir las EP. Para ello, se intenta mejorar el AMC y evitar o controlar la infección bronquial. Los tratamientos habituales de un paciente con FQ incluyen, entre otros, los que se exponen a continuación.
Fisioterapia respiratoria
Es un pilar fundamental del tratamiento de la FQ(2,7). Existen diferentes técnicas, aunque ninguna ha demostrado ser mejor que las otras. La elección dependerá, por tanto, de la edad y las preferencias del paciente, así como de la experiencia del equipo rehabilitador. En general, se aconseja realizar la fisioterapia dos veces al día, aunque se puede incrementar la frecuencia en las EP o en situaciones de deterioro clínico(7).
Suero salino hipertónico al 7% inhalado
Mejora el AMC, hidratando las secreciones respiratorias y facilitando su drenaje, por lo que disminuye las EP(3). Se utiliza antes de la fisioterapia respiratoria(7). Su uso se inicia lo antes posible, tras el diagnóstico de FQ. Se debe administrar previamente un broncodilatador y tener precaución en pacientes con hiperreactividad bronquial.
DNasa recombinante inhalada
Fue el primer fármaco específicamente autorizado para el tratamiento de la FQ. Actúa degradando el ADN de neutrófilos y bacterias presente en las secreciones respiratorias, disminuyendo su viscosidad y favoreciendo su aclaramiento(10).
Antibióticos inhalados
La administración de antibióticos por vía inhalada permite alcanzar concentraciones elevadas a nivel bronquial, minimizando la absorción sistémica y, por tanto, los efectos secundarios(11). Existen formulaciones específicas para nebulización de tobramicina, colistimetato sódico y aztreonam lisina, así como presentaciones de tobramicina y colistimetato de sodio en polvo seco. Entre los antibióticos nebulizados en desarrollo, se encuentran: amikacina, vancomicina y levofloxacino. También pueden utilizarse, por vía inhalada, formulaciones intravenosas de antibiótico, asegurando previamente que el paciente lo tolera adecuadamente. Los antibióticos nebulizados se utilizan para el control de la infección bronquial crónica(7) y en regímenes erradicadores de infecciones por P. aeruginosa, el complejo Burkholderia cepacia o micobacterias no tuberculosas.
Broncodilatadores
Los pacientes con FQ tienen más hiperreactividad bronquial que la población general, por lo que podría estar justificado su uso si la clínica o la espirometría sugieren un beneficio clínico. También se emplean antes de las nebulizaciones para mejorar su tolerancia(7).
Antiinflamatorios
El ibuprofeno a altas dosis ha demostrado beneficio clínico a largo plazo, pero con efectos secundarios importantes y con necesidad de una monitorización estrecha, por lo que no se usa de forma habitual. Los corticoides sistémicos no producen beneficios a largo plazo y tienen muchos efectos secundarios, por lo que no se aconseja su uso(3). Los corticoides inhalados no han demostrado beneficios a largo plazo, por lo que están sólo indicados en pacientes que tienen asma asociada a FQ(7). La azitromicina administrada de forma crónica (10 mg/kg, en pacientes con peso <40 kg, 250-500 mg, en pacientes >40 kg), tres días por semana, tienen un efecto inmunomodulador y disminuyen las EP, sobre todo en pacientes con infección bronquial crónica por P. aeruginosa(7,11).
Oxigenoterapia y apoyo ventilatorio
La oxigenoterapia nocturna está indicada cuando se demuestre una hipoxia nocturna, lo que debería evaluarse cuando el FEV1 del paciente cae por debajo del 50%(7). Si el paciente tiene hipoxia o disnea diurna, se debe indicar oxigenoterapia continua (o el mayor número de horas que tolere). La ventilación mecánica no invasiva ha demostrado su eficacia en pacientes del injerto con insuficiencia respiratoria crónica y puede ser necesaria durante las EP en algunos pacientes(10).
Trasplante pulmonar
Es la única opción terapéutica cuando la función pulmonar está muy afectada y la calidad de vida muy deteriorada; aunque es un tratamiento no exento de complicaciones y con una supervivencia del injerto en torno al 60% a los 5 años(10). Se indica en pacientes con enfermedad pulmonar avanzada sintomática, con una esperanza de vida teórica inferior a 2 años y en ausencia de contraindicaciones.
Tratamiento con moduladores de CFTR
El descubrimiento de la familia de los moduladores, potenciadores y correctores de CFTR, ha supuesto un cambio muy significativo en el tratamiento de los pacientes con FQ, ya que, por primera vez, se dispone de fármacos que modifican la fisiopatología de la enfermedad. El uso de estos fármacos, de una manera precoz, tendría el potencial de corregir la función de CFTR y evitar toda la cascada fisiopatológica de esta enfermedad (Fig.1).
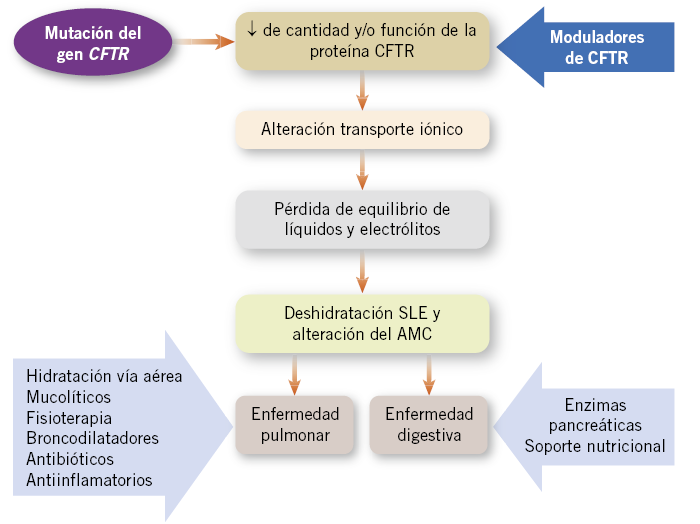
Figura 1. Esquema de la cascada fisiopatológica de la fibrosis quística. Los tratamientos habituales se dirigen a las complicaciones de la enfermedad, al final de la cascada fisiopatológica. Los nuevos tratamientos moduladores de CFTR (potenciadores y correctores) actúan al inicio de la cascada fisiopatológica y, potencialmente, podrían evitar o disminuir las consecuencias típicas de la enfermedad. CFTR: regulador de la conductancia transmembrana de la fibrosis quística; SLE: superficie líquida del epitelio y AMC: aclaramiento mucociliar.
El primer fármaco desarrollado fue ivacaftor (Kalydeco®), un potenciador capaz de aumentar la probabilidad de apertura del canal CFTR, que en pacientes con mutaciones de clase III ha demostrado mejorar la función pulmonar, el estado nutricional y disminuir las EP(17). Actualmente, está indicado en pacientes con mutaciones de clase III, que en España suponen <5% de los pacientes, a partir de los 4 meses de edad.
La siguiente generación de fármacos, los correctores, son capaces de corregir el defecto de plegamiento de la proteína y consiguen que se expresen canales CFTR en la superficie de la célula epitelial, aunque el canal resultante tiene defectos de apertura y estabilidad; su uso en monoterapia no consiguió ningún efecto clínico significativo. Sin embargo, la combinación de un corrector con un potenciador, sí consiguió efectos clínicos relevantes en pacientes con FQ homocigotos para la mutación Phe.508del. Los ensayos clínicos de las combinaciones lumacaftor-ivacaftor y tezacaftor-ivacaftor han demostrado mejorías del FEV1 y ganancia de peso, pero mucho más modestas que las obtenidas por ivacaftor(17). En la actualidad, lumacaftor-ivacaftor (Orkambi®) está aprobado para pacientes mayores de 6 años con FQ , en España, homocigotos para la mutación Phe.508del, y tezacaftor-ivacaftor (Symkevi®) para pacientes mayores de 12 años homocigotos para la mutación Phe.508del o la combinación de Phe.508del con alguna mutación con función residual.
La siguiente combinación, recientemente aprobada por la FDA (Food and Drug Administration), la EMA (Agencia Europea del Medicamento), y la AEMPS (Agencia Española del Medicamento y Productos Sanitarios) para pacientes > de 12 años, incluye la combinación ivacaftor-tezacaftor y un tercer corrector (elexacaftor), cuya efectividad ha sido demostrada en pacientes FQ con una sola mutación Phe.508del(18). Los resultados clínicos de esta triple combinación superan de manera notable los conseguidos por las combinaciones anteriores y, además, es adecuada para tratar aproximadamente al 90% de los enfermos con FQ (75% en España) que portan, al menos, una mutación Phe.508del. Se espera que la combinación ivacaftor-tezacaftor-elexacaftor (Kaftrio®) pueda estar disponible es España durante 2021.
Además de los moduladores descritos, están en desarrollo nuevos correctores y potenciadores, así como estabilizadores y amplificadores de CFTR, que ampliarán el escaso arsenal terapéutico del que disponemos para el tratamiento de la FQ(19). Sin embargo, en el momento actual, aproximadamente un 10% de los pacientes no son susceptibles de recibir tratamiento con moduladores, debido a que no son portadores de ninguna mutación que responda a estos fármacos. El futuro del tratamiento de estos pacientes podría pasar por agentes para el tratamiento de otras clases de mutaciones, el tratamiento con la tecnología de ARNm o la tecnología de edición genética CRISPR, que tiene el potencial de corregir el defecto genético y dar una solución definitiva a la enfermedad(19).
Tratamiento de la exacerbación pulmonar
El uso de antibióticos es fundamental en el tratamiento de las exacerbaciones respiratorias en la fibrosis quística.
Las EP requieren tratamiento antibiótico a dosis elevadas y durante más tiempo que en la población general (2-3 semanas)(2). El tratamiento debe guiarse, siempre que se pueda, por el resultado microbiológico con estudio de sensibilidad in vitrode los cultivos previos del paciente(7). En EP leves o moderadas puede emplearse la vía oral. En los pacientes sin infección por P. aeruginosa, suele emplearse amoxicilina-ácido clavulánico o trimetoprim-sulfametoxazol; en caso de infección por P. aeruginosa, se empleará ciprofloxacino, independientemente de la edad, ya que su uso se ha demostrado seguro en Pediatría. Las EP graves requieren el uso de antibióticos intravenosos: amoxicilina-ácido clavulánico y/o trimetoprim-sulfametoxazol; en caso de infección por P. aeruginosa, se usará ceftazidima más un aminoglucósido(13). Con la edad, se hacen más frecuentes las infecciones por bacterias multirresistentes, lo que implicaría el uso de otros antibióticos diferentes. Los antibióticos orales más usados en FQ se resumen en la tabla IV.

Complicaciones respiratorias de la fibrosis quística
Las complicaciones no infecciosas de la fibrosis quística influyen negativamente en su pronóstico, por lo que deben diagnosticarse y tratarse de manera adecuada.
Las complicaciones más frecuentes son: neumotórax, atelectasia, hemoptisis, hipertensión pulmonar y desarrollo de insuficiencia respiratoria crónica.
Neumotórax
Ocurre en menos del 5% de los pacientes con FQ, habitualmente, en situaciones de enfermedad pulmonar avanzada. Se manifiesta como: dificultad respiratoria y/o dolor torácico pleurítico punzante de aparición brusca, irradiado hacia el hombro ipsilateral, con signos de dificultad respiratoria en los de gran tamaño. La radiografía de tórax en bipedestación y espiración, suele demostrar una línea de la pleura visceral y la ausencia de vasos pulmonares en la zona afectada, con desplazamiento contralateral de la tráquea y del mediastino, y atelectasia del pulmón afectado en los casos más graves. El tratamiento es la oxigenoterapia y analgesia en los casos leves, y la colocación de un tubo de drenaje pleural en los graves. La pleurodesis puede evitar recurrencias, pero puede complicar un futuro trasplante pulmonar(20).
Atelectasia
Es el colapso o pérdida de volumen pulmonar, habitualmente de un segmento o lóbulo completo, que se produce por una obstrucción de la luz bronquial. Con frecuencia, es asintomática y se descubre incidentalmente en una prueba de imagen indicada por otro motivo. La exploración física puede ser, por tanto, completamente normal. En otras ocasiones, se manifiesta como una EP. La radiografía de tórax es diagnóstica, en la mayoría de los casos. La TC de tórax es más sensible para detectar atelectasias pequeñas, en ocasiones, de dudosa repercusión clínica(2). El tratamiento incluye: uso de antibióticos intravenosos, broncodilatadores, mucolíticos (DNasa) y fisioterapia respiratoria intensa. Puede ser necesario realizar una fibrobroncoscopia con aspiración de secreciones e instilación local de DNasa, o suero salino hipertónico. En casos refractarios muy sintomáticos, puede plantearse la lobectomía quirúrgica.
Hemoptisis
La hemoptisis afecta generalmente a pacientes con enfermedad pulmonar moderada o grave. Se produce por sangrado de arterias bronquiales aberrantes, que aparecen en el contexto de una enfermedad pulmonar progresiva, que destruye y altera la arquitectura normal del pulmón y la vía aérea. La hepatopatía y el déficit de vitamina K favorecen su aparición. La hemoptisis leve (<5 mL) se manifiesta como esputos mezclados con sangre roja, casi siempre coincidiendo con una EP. La hemoptisis masiva (>240 mL) puede acompañarse de obstrucción de la vía aérea, insuficiencia respiratoria e inestabilidad hemodinámica significativa. El diagnóstico de la hemoptisis es clínico y no necesita de pruebas complementarias. En hemoptisis masivas, es necesario realizar una angio-TC para detectar el origen del sangrado. El tratamiento antibiótico de la EP suele detener la hemoptisis, incluso cuando esta es significativa. En casos graves, conviene suspender el tratamiento inhalado y la fisioterapia durante 2-3 días, para evitar el resangrado. En casos refractarios, está indicada la embolización de la arteria bronquial sangrante(7,10,20).
Aspergilosis broncopulmonar alérgica (ABPA)
La ABPA se produce como resultado de una compleja reacción de hipersensibilidad, tanto tipo I como III, en respuesta a la presencia en la vía aérea de Aspergillus fumigatus. Provoca síntomas similares a una EP, pero sin respuesta al tratamiento antibiótico habitual. Otras veces, su curso es más insidioso, con un deterioro subagudo y con mala respuesta al tratamiento. Es característica la emisión de esputos de color marronáceo, acompañados o no de hemoptisis. Su diagnóstico es complicado y precisa cumplir una serie de criterios, entre ellos, la elevación de la IgE por encima de 1.000 kU/L(14).
Para el tratamiento, se utilizan corticoides sistémicos (prednisona a 0,5-2 mg/kg/día), durante 1-2 semanas, con reducción lenta posterior hasta suspenderla en 2-3 meses. Como alternativa, se han empleado pulsos de metilprednisolona (10-30 mg/kg/día), durante 3 días, repetidos a intervalos de 4 semanas. Los antifúngicos (itraconazol 2,5 mg/kg/12 horas, máximo 200 mg/dosis), se emplean para reducir la carga de Aspergillus en el árbol bronquial. También, puede emplearse voriconazol. Otras opciones terapéuticas, fuera de indicación, son la anfotericina B nebulizada o el omalizumab(7,10).
Fallo respiratorio crónico hipoxémico asociado o no a hipercapnia
Cuando la enfermedad pulmonar avanza (FEV1 menor del 50%), la destrucción tisular y la consiguiente alteración de la ventilación-perfusión conducen al fallo respiratorio crónico hipoxémico, asociado o no a hipercapnia. Se debe emplear oxígeno suplementario cuando la saturación transcutánea de O2 es menor del 88% durante el ejercicio. La cefalea matutina, la disnea o la mala calidad del sueño pueden ser síntomas de hipercapnia, que debe ser estudiada con gasometría para valorar la necesidad de ventilación mecánica no invasiva.
Hipertensión pulmonar
Aparece en pacientes con enfermedad pulmonar grave, por fibrosis de la íntima y vascularización de las arteriolas pulmonares. Se asocia a mayor riesgo de mortalidad y sus síntomas, muchas veces, se superponen a los de la propia enfermedad pulmonar. Por este motivo, debe buscarse activamente, mediante ecocardiografía transtorácica o, en caso necesario, un cateterismo con medición directa de las presiones pulmonares. El tratamiento es el oxígeno suplementario, aunque hay casos descritos de buena evolución con inhibidores de la fosfodiesterasa y prostaciclinas inhaladas(10,20).
Función del pediatra de Atención Primaria
La función del pediatra de Atención Primaria en la FQ es limitada, ya que los pacientes suelen ser vistos en Unidades Multidisciplinares que se ocupan de casi todos los aspectos relacionados con su salud. En el paciente no diagnosticado, el pediatra debe mantener un alto índice de sospecha ante síntomas de: tos húmeda crónica, malabsorción, fallo de medro y episodios de deshidratación grave. En el paciente ya diagnosticado, desde Atención Primaria deben ofrecerse las inmunizaciones correspondientes al calendario vigente, así como, anualmente, la vacunación frente a la gripe. Además, en regiones sin unidades de referencia, el pediatra de Atención Primaria debe ser quien realice la primera valoración del paciente con síntomas, instaure el tratamiento inicial y contacte con la Unidad de Referencia de Fibrosis Quística.
Bibliografía
Los asteriscos muestran el interés del artículo a juicio de los autores.
1. Goetz D, Ren CL. Review of Cystic Fibrosis. Pediatr Ann. 2019; 48: e154-e61. doi: 10.3928/19382359-20190327-01.
2.*** Bell SC, Mall MA, Gutiérrez H, Macek M, Madge S, Davies JC, et al. The future of cystic fibrosis care: a global perspective. Lancet Respir Med. 2020; 8: 65-124. doi: 10.1016/s2213-2600(19)30337-6.
3. De Boeck K, Amaral MD. Progress in therapies for cystic fibrosis. Lancet Respir Med. 2016; 4: 662-74. doi: 10.1016/S2213-2600(16)00023-0.
4. Pagin A, Sermet-Gaudelus I, Burgel PR. Genetic diagnosis in practice: From cystic fibrosis to CFTR-related disorders. Arch Pediatr. 2020; 27: eS25-eS29. doi: 10.1016/s0929-693x(20)30047-6.
5. Zolin A, Orenti A, Naehrlich L, Jung A, van Rens J, Fox A, et al. ECFS Patient Registry Annual Data Report. 2018. Acceso el 30 de diciembre de 2020. Disponible en: https://www.ecfs.eu/projects/ecfs-patient-registry/annual-reports.
6. Balfour-Lynn IM, King JA. CFTR modulator therapies – Effect on life expectancy in people with cystic fibrosis. Paediatr Respir Rev. 2020. doi: 10.1016/j.prrv.2020.05.002.
7.** Castellani C, Duff AJA, Bell SC, Heijerman HGM, Munck A, Ratjen F, et al. ECFS best practice guidelines: the 2018 revision. Journal of Cystic Fibrosis. 2018; 17: 153-78. doi: 10.1016/j.jcf.2018.02.006.
8. Naehrlich L. The Changing Face of Cystic Fibrosis and Its Implications for Screening. Int J Neonatal Screen. 2020; 6: 54. doi: 10.3390/ijns6030054.
9. Roesch EA, Nichols DP, Chmiel JF. Inflammation in cystic fibrosis: An update. Pediatr Pulmonol. 2018; 53: S30-S50. doi: 10.1002/ppul.24129.
10.*** Turcios NL. Cystic Fibrosis Lung Disease: An Overview. Respir Care. 2020; 65: 233-51. doi: 10.4187/respcare.06697.
11. Ratjen F, Bell SC, Rowe SM, Goss CH, Quittner AL, Bush A. Cystic fibrosis. Nat Rev Dis Primers. 2015; 1: 15010. doi: 10.1038/nrdp.2015.10.
12. De Dios Caballero J, Del Campo R, Royuela A, Solé A, Máiz L, Olveira C, et al. Bronchopulmonary infection-colonization patterns in Spanish cystic fibrosis patients: Results from a national multicenter study. J Cyst Fibros. 2016; 15: 357-65. doi: 10.1016/j.jcf.2015.09.004.
13.** Cantón R, Máiz L, Escribano A, Olveira C, Oliver A, Asensio O, et al. Consenso español para la prevención y el tratamiento de la infección bronquial por Pseudomonas aeruginosa en el paciente con fibrosis quística. Arch Bronconeumol. 2015; 51: 140-50. doi: 10.1016/j.arbres.2014.09.021.
14. Lattanzi C, Messina G, Fainardi V, Tripodi MC, Pisi G, Esposito S. Allergic Bronchopulmonary Aspergillosis in Children with Cystic Fibrosis: An Update on the Newest Diagnostic Tools and Therapeutic Approaches. 2020; 9. doi: 10.3390/pathogens9090716.
15. Stanojevic S, Davis SD, Retsch-Bogart G, Webster H, Davis M, Johnson RC, et al. Progression of Lung Disease in Preschool Patients with Cystic Fibrosis. 2017; 195: 1216-25. doi: 10.1164/rccm.201610-2158OC.
16. Hota P, Madan R. Cystic Fibrosis from Childhood to Adulthood: What Is New in Imaging Assessment? Radiol Clin North Am. 2020; 58: 475-86. doi: 10.1016/j.rcl.2019.12.003.
17.*** Lopes-Pacheco M. CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine. Front Pharmacol. 2019; 10: 1662. doi: 10.3389/fphar.2019.01662.
18. Keating D, Marigowda G, Burr L, Daines C, Mall MA, McKone EF, et al. VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N Engl J Med. 2018; 379: 1612-20. doi: 10.1056/NEJMoa1807120.
19. Pranke I, Golec A, Hinzpeter A, Edelman A, Sermet-Gaudelus I. Emerging Therapeutic Approaches for Cystic Fibrosis. From Gene Editing to Personalized Medicine. Front Pharmacol. 2019; 10: 121. doi: 10.3389/fphar.2019.00121.
20.*** García B, Flume PA. Pulmonary Complications of Cystic Fibrosis. Semin Respir Crit Care Med. 2019; 40: 804-09. doi: 10.1055/s-0039-1697639.
Bibliografía recomendada
- Bell SC, Mall MA, Gutiérrez H, Macek M, Madge S, Davies JC, et al. The future of cystic fibrosis care: a global perspective. Lancet Respir Med. 2020; 8: 65-124. doi: 10.1016/s2213-2600(19)30337-6.
Excelente y completa revisión actualizada de la fibrosis quística, que incluye: desde el diagnóstico y el tratamiento actual, hasta los tratamientos futuros, la realidad de la fibrosis quística en los países en vías de desarrollo o el papel de las asociaciones de pacientes.
- Castellani C, Duff AJA, Bell SC, Heijerman HGM, Munck A, Ratjen F, et al. ECFS best practice guidelines: the 2018 revision. Journal of Cystic Fibrosis. 2018; 17: 153-78. doi: 10.1016/j.jcf.2018.02.006.
Actualización más reciente de las guías de práctica clínica de la Sociedad Europea de Fibrosis Quística, que abarca todos los aspectos de la enfermedad.
- García B, Flume PA. Pulmonary Complications of Cystic Fibrosis. Semin Respir Crit Care Med. 2019; 40: 804-09. doi: 10.1055/s-0039-1697639.
Artículo de revisión sobre las complicaciones pulmonares más importantes de la fibrosis quística, con los tratamientos recomendados para cada una de ellas.
- Lopes-Pacheco M. CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine. Front Pharmacol. 2019; 10: 1662. doi: 10.3389/fphar.2019.01662.
Artículo de revisión que repasa los nuevos tratamientos para la fibrosis quística, así como las posibles opciones terapéuticas a medio plazo.
- Turcios NL. Cystic Fibrosis Lung Disease: An Overview. Respir Care. 2020; 65: 233-51. doi: 10.4187/respcare.06697.
Artículo de revisión que repasa todo el espectro de manifestaciones respiratorias de la fibrosis quística, incluyendo sus complicaciones.
| Caso clínico |
|
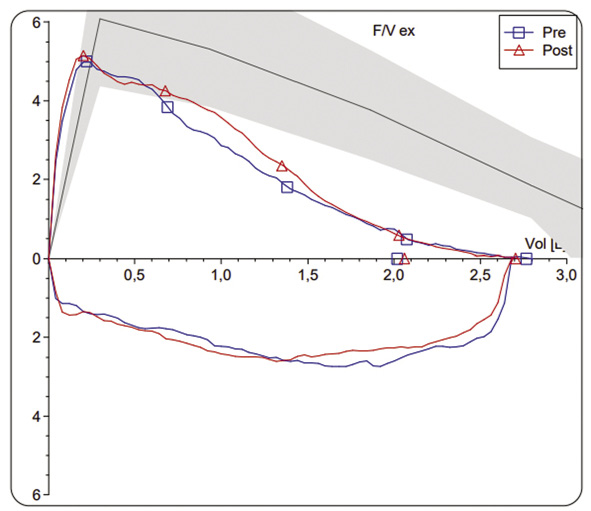
Anamnesis Varón de 9 años de edad, con diagnóstico de fibrosis quística (FQ) por retraso ponderal y cuadro de deshidratación hiponatrémica e hipoclorémica a los 9 meses de vida. Acude a consulta por clínica de tos con expectoración escasa, de unas 3 semanas de evolución, y aparición de disnea con los esfuerzos moderados. No ha tenido fiebre ni pérdida de apetito. No se describen cambios significativos en el esputo. Antecedentes personales Embarazo y parto normales. Escasa ganancia ponderal y deposiciones pastosas, frecuentes y abundantes, hasta el diagnóstico de FQ a los 9 meses de vida. Cribado neonatal no instaurado en su Comunidad Autónoma en el momento del nacimiento. Genotipo Gly542X (G542X) / Asn1303Lys (N1303K). Cloro en sudor 99 y 107 mEq/L. Insuficiencia pancreática. Infección bronquial crónica por Staphylococcus aureus sensible a la meticilina e intermitente por Pseudomonas aeruginosa (último cultivo positivo hace 9 meses). Afectación pulmonar leve con FEV1 del 85% sobre el predicho. Tratamiento habitual: enzimas pancreáticas en las comidas, suplementos vitamínicos. Nebulizaciones con suero salino hipertónico 7% cada 12 horas y fisioterapia respiratoria 2 veces al día. Último antibiótico inhalado (tobramicina) finalizado hace 3 meses. Exploración física Peso: 24,8 kg (p20). Talla: 122 cm (p4). Temperatura: 36,4ºC. SatO2: 97%. Buen estado general. Sin lesiones cutáneas. Sin signos de dificultad respiratoria. Auscultación cardiaca: rítmica sin soplos. Auscultación pulmonar: buena ventilación bilateral, con crepitantes dispersos. Abdomen blando, no doloroso. ORL: normal. Neurológico normal. Exámenes complementarios Espirometría basal y tras broncodilatador: FEV1 2,02 L (z-score -3,08, 62%); FVC: 2,77 (z-score -2,26, 74%); FEV1/FVC 73%. FEV1 post-broncodilatador 2,06 (+2%) (Fig. 2).
Figura 2. Se recoge esputo para cultivo microbiológico con estudio de sensibilidad in vitro. Evolución Se diagnostica de exacerbación pulmonar leve y se inicia tratamiento con ciprofloxacino oral. Acude a las 2 semanas sin mejoría clínica ni espirométrica, por lo que se decide realizar radiografía de tórax (Fig. 3) e ingreso hospitalario para tratamiento antibiótico intravenoso.
Figura 3. Resultado del cultivo: Staphylococcus aureus sensible a la meticilina. Aspergillus fumigatus. Análisis de sangre: hemograma y bioquímica normales con PCR: 5 mg/L. IgE total: 27 kU/L.
|


 New approach in the treatment of children with asthma
New approach in the treatment of children with asthma