|
| Regreso a las bases |
D. Rodríguez Álvarez, M.D. del Río García
Servicio de Cuidados Intensivos Pediátricos. Hospital Universitario La Paz. Madrid
| Resumen
La reanimación cardiopulmonar (RCP) pediátrica es una técnica que puede salvar vidas. En este artículo se describen las actuaciones básicas a realizar ante un niño que se encuentre en situación de parada cardiorrespiratoria (PCR). La secuencia de actuación se puede resumir en los siguientes pasos: seguridad, estimular, pedir ayuda, abrir vía aérea, valoración de la ventilación, ventilación, valoración de la circulación y compresiones torácicas. Además, en este tema se incluyen: el manejo de una obstrucción de la vía aérea por un cuerpo extraño (OVACE) y la utilización del desfibrilador semiautomático (DESA). Por último, se resaltan los cambios que existen entre estas recomendaciones y las anteriores de 2015. |
| Abstract
Pediatric cardiopulmonary resuscitation (CPR) is a life-saving technique. This article outlines the basic actions to be taken when dealing with a child experiencing cardiopulmonary arrest (CPA). The sequence of steps can be summarized as follows: safety, stimulation, calling for help, opening the airway, assessing ventilation, providing ventilation, assessing circulation, and chest compressions. Additionally, this topic includes: managing a foreign body airway obstruction (FBAO) and using an automated external defibrillator (AED). Finally, it highlights the changes between these recommendations and the previous ones from 2015. |
Palabras clave: Reanimación cardiopulmonar básica; Parada cardiorrespiratoria; Soporte vital básico; Obstrucción de la vía aérea; Desfibrilador externo semiautomático.
Key words: Basic cardiopulmonary resuscitation; Cardiorespiratory arrest; Basic life support; Airway obstruction; Semiautomatic external defibrillator.
Pediatr Integral 2024; XXVIII (1): 58 – 64
OBJETIVOS
• Reconocer una parada cardiorrespiratoria y realizar una evaluación rápida en situaciones de emergencia.
• Conocer los pasos de la reanimación cardiopulmonar (RCP) pediátrica básica, desde la activación del sistema de emergencias hasta la realización de compresiones torácicas y ventilaciones.
• Adaptar la RCP pediátrica según la edad y el tamaño del niño, reconociendo las diferencias entre lactantes y niños.
• Reconocer un episodio de obstrucción de la vía respiratoria superior y saber llevar a cabo las maniobras de desobstrucción.
• Integrar el uso del desfibrilador externo semiautomático (DESA) en la RCP pediátrica básica.
Reanimación cardiopulmonar básica y semiavanzada
Introducción
La reanimación cardiopulmonar (RCP) pediátrica es un conjunto de conocimientos y habilidades esenciales para el pediatra de Atención Primaria. Cuando un niño experimenta una parada cardiorrespiratoria (PCR), cada segundo cuenta, y saber cómo realizar adecuadamente la RCP puede ser la clave para salvar su vida y evitar daños cerebrales irreversibles(1). Es de vital importancia ser capaz de identificar rápidamente las situaciones de PCR en niños y conocer las maniobras de RCP básica. Así, podremos iniciar la reanimación de manera oportuna y aumentar la probabilidad de supervivencia en estos pacientes. En esta sección de Regreso a las Bases, exploraremos en detalle los fundamentos y recomendaciones actuales de la RCP pediátrica, con el objetivo de ofrecer la información necesaria para actuar de manera efectiva y segura en una situación de PCR pediátrica.
Epidemiología
En las últimas décadas, se ha observado una disminución en la incidencia de PCR extrahospitalaria. En el caso de los niños, un estudio en EE.UU. estima que la incidencia se encuentra en un rango aproximado de 0,6 a 2,1 casos por cada 100.000 personas al año, siendo más alta en el grupo de menores de 1 año(2).
En cuanto a la supervivencia, es significativamente menor (11 %) en los casos de PCR extrahospitalaria en comparación con la intrahospitalaria (41 %). Además, la supervivencia también varía según la edad del paciente. En el grupo de adolescentes se observa una tasa de supervivencia del 19,2 %; mientras que en el grupo de niños menores de 1 año, la tasa de supervivencia es del 6,7 %(3).
La principal causa de PCR en niños es el fallo respiratorio. Por lo tanto, las prioridades durante la realización de la RCP básica son asegurar una vía aérea abierta y administrar oxígeno de manera inmediata a través de las insuflaciones del reanimador.
En cuanto a las PCR de origen cardiaco, la bradicardia grave que progresa hacia la actividad eléctrica sin pulso y posterior asistolia es la arritmia más común en la parada cardíaca pediátrica, siendo por ello fundamental realizar una RCP básica de alta calidad. Sin embargo, a medida que aumenta la edad, es más frecuente la fibrilación ventricular, que se manifiesta como PCR con colapso súbito e inesperado, por lo que resulta crucial disponer de un DESA cuanto antes y saber usarlo adecuadamente.
Conceptos previos
En RCP pediátrica, se utiliza el término “lactantes” para referirse a los niños menores de 1 año, excluyendo a los recién nacidos (con recomendaciones específicas de reanimación tras el parto), y se utiliza el término “niños” para aquellos con edades comprendidas entre 1 año y la pubertad. No es necesario definir específicamente el inicio de la pubertad. Si el reanimador determina que la víctima es un niño, se deben aplicar las recomendaciones específicas para la RCP pediátrica.
Recomendaciones
Este artículo ha sido elaborado siguiendo las recomendaciones publicadas por el European Resuscitation Council (ERC) en el año 2021(4), adaptadas por el Grupo Español de Reanimación Cardiopulmonar Pediátrica y Neonatal (GERCPPYN)(5) y basadas en las directrices emitidas por el International Liaison Committee on Resuscitation (ILCOR) en 2020(6). Recientemente, se han publicado las recomendaciones ILCOR 2023(7), en las que no se sugieren modificaciones para la secuencia de RCP básica pediátrica. En un apartado posterior, se destacará de manera especial los cambios relevantes con respecto a las recomendaciones anteriores de 2015(8).
Reanimación cardiopulmonar básica
Una PCR se define como: la interrupción brusca, inesperada y potencialmente reversible de la respiración y la circulación espontáneas. La RCP básica comprende la identificación del individuo que experimenta una PCR, la notificación a los servicios de emergencia médica (SEM) y la asistencia en las funciones respiratorias y circulatorias hasta que el paciente pueda recibir atención sanitaria más especializada. Además del DESA, no se requiere la utilización de más equipamiento específico. Se indica la realización de RCP básica en cualquier niño que no responde a estímulos y presenta alteración en la función respiratoria. Debe iniciarse lo más pronto posible, idealmente por aquellas personas que presencien la emergencia.
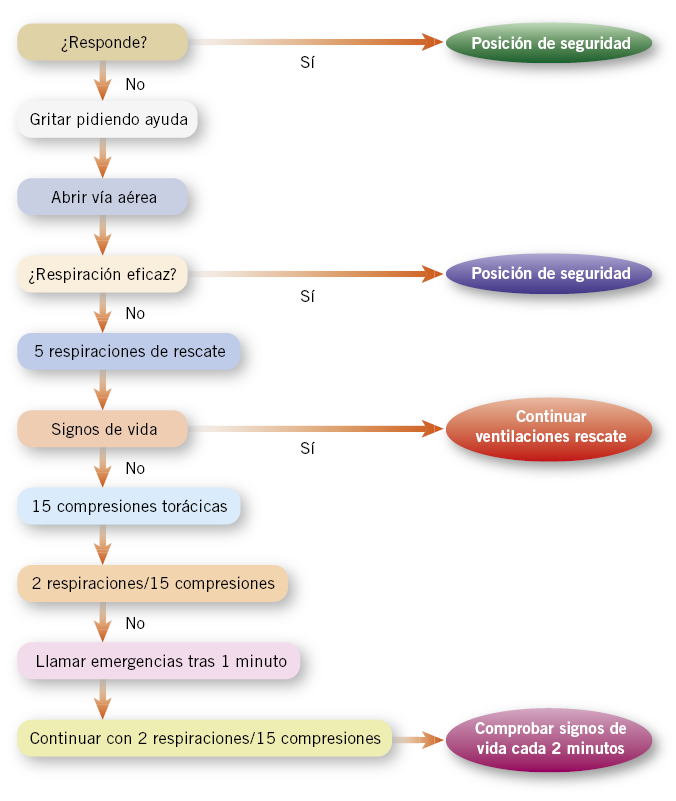
La secuencia de actuaciones en la RCP básica se resume en la figura 1 y se detalla a continuación:
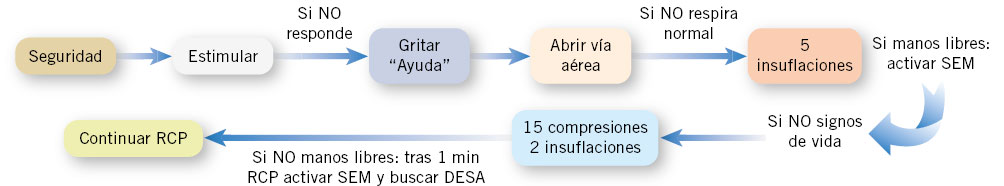
Figura 1. Reanimación cardiopulmonar (RCP) básica con un reanimador. SEM: Servicios de Asistencia Médica; DESA: desfibrilador semiautomático.
1. Seguridad:
• Reconocer rápidamente el escenario y la situación donde se desarrolla la PCR para garantizar la seguridad, tanto de la víctima como del equipo reanimador. Por ejemplo, ante un accidente de tráfico, la primera medida será controlar el tráfico para proteger y protegerse.
• Movilizar a la víctima a un lugar seguro, si fuese necesario y posible.
• Utilizar guantes y otros métodos de barrera, si se dispone de ellos. Todos los líquidos corporales (sangre, saliva, vómito…) deben considerarse como potencialmente infecciosos.
2. Estimular:
• Establecer el grado de respuesta del niño a estímulos verbales, gritándole “¿cómo te encuentras?” o llamándole por su nombre si se conociese, y a estímulos dolorosos, presionando en el reborde supraorbitario, por ejemplo. En los niños en los que se sospeche una lesión cervical, estos estímulos se harán con precaución, inmovilizando el cuello.
• Se considerará que responde, si el niño se mueve, llora, grita o habla. En ese caso, se le dejará en la posición en que se encuentra, se pedirá ayuda y se revalorará periódicamente.
• Si no respondiese, se continuará con el algoritmo de RCP básica.
3. Gritar ayuda:
• Gritar pidiendo ayuda a los transeúntes del lugar.
• Si solo hubiese un reanimador, este deberá comenzar la RCP.
• Con dos o más reanimadores, el segundo reanimador, si dispone de teléfono, deberá llamar al 112 (número de emergencias en la Unión Europea), preferiblemente usando el modo “manos libres”. Durante la llamada, debe indicarse: el lugar donde se está llevando a cabo la reanimación, la edad aproximada del paciente, qué ha sucedido (en el caso de una PCR presenciada) y qué maniobras se están realizando. Es posible recibir indicaciones telefónicas sobre cómo continuar con las actuaciones de reanimación.
• Colocar al paciente en decúbito supino sobre una superficie plana y firme, de manera que se puedan realizar las maniobras de reanimación con la máxima eficacia.
4. Abrir vía aérea (A):
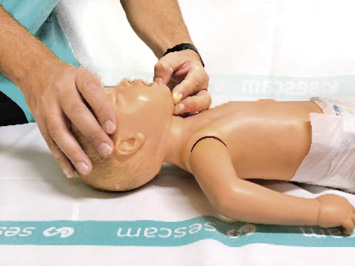
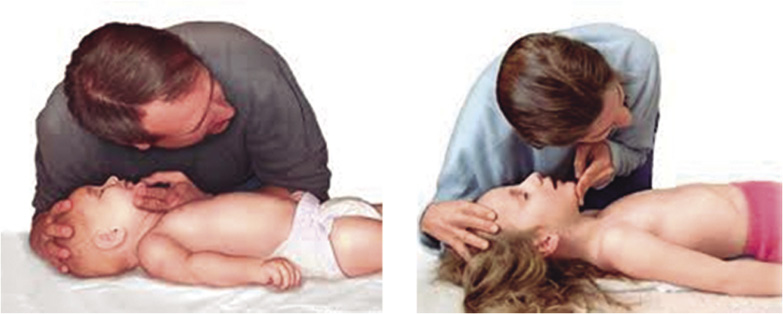
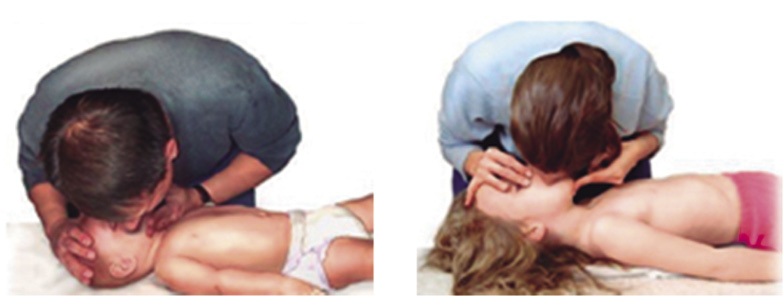
• Abrir la vía aérea, utilizando la maniobra de elección frente-mentón (Fig. 2).

Figura 2. Maniobra frente-mentón.
Situándonos a un lado del niño, colocaremos la palma de una mano en la frente del niño y los dedos de la otra mano sobre su mandíbula, extendiendo el cuello hasta una posición neutra en los lactantes (posición de “olfateo”) y hasta una ligera hiperextensión en los niños. Se deberá evitar la flexión y la hiperextensión cervical excesiva, así como la presión en las partes blandas mandibulares, ya que estas acciones pueden provocar el colapso de la vía aérea.
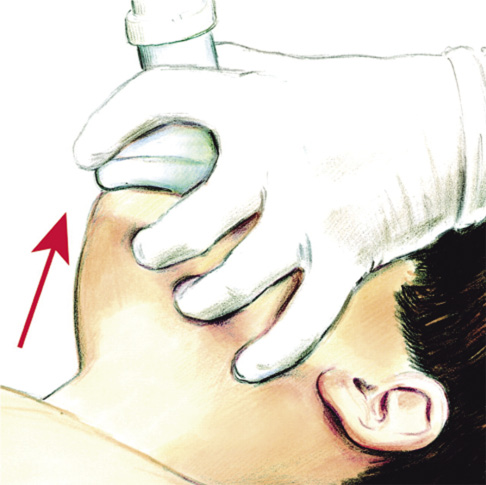
• Si existe riesgo de lesión cervical, se recomiendan las maniobras de tracción mandibular y de elevación mandibular:
– La maniobra de tracción mandibular (Fig. 3A) se realiza colocando la palma de una mano en la frente del niño, bloqueando la extensión del cuello, e introduciendo el dedo pulgar de la otra mano en la boca, detrás de los incisivos centrales inferiores, mientras que los dedos índice y medio se sitúan debajo del mentón, formando una pinza para traccionar la mandíbula.
– La maniobra de elevación mandibular (Fig. 3B) la realizaremos colocándonos detrás del niño. Posicionaremos las manos a ambos lados de la cabeza. Los dedos pulgares deberán situarse en las mejillas del niño, mientras que los dedos índice y corazón se ubicarán en ambos ángulos mandibulares, elevándola la mandíbula hacia arriba.

Figura 3. A. Maniobra de tracción mandibular. B. Maniobra de elevación mandibular.
• Explorar el interior de la boca en busca de cuerpos extraños.
• En caso se observar un cuerpo extraño, se procederá a extraerlo mediante barrido lateral con un solo dedo, con extrema precaución para no hacer avanzar el cuerpo extraño en la vía aérea superior. No se debe realizar nunca un barrido a ciegas de la cavidad bucal.
5. Valoración de la ventilación (B):
• Tras la apertura de la vía aérea, hay que comprobar si el niño realiza respiraciones efectivas espontáneas. Para ello, el reanimador colocará su mejilla cerca de la boca y nariz del niño, durante un máximo de 10 segundos, y, mediante la maniobra “ver, oír, sentir”, determinará si el niño respira o no. “Ver”: observará si el tórax se mueve; “oír”: detectará si existen ruidos sugestivos del paso de aire por las vías respiratorias; “sentir”: notará aire en su mejilla si el paciente exhalase.
• La población general puede valorar la existencia de respiración poniendo una mano sobre el tórax o el abdomen para comprobar si estos se mueven.
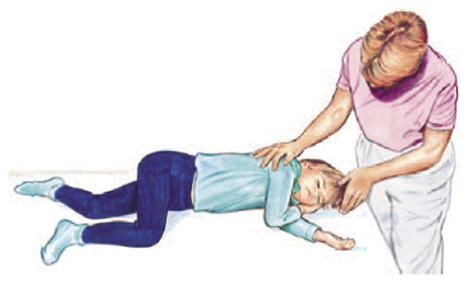
• Si el niño respira, se colocará en posición lateral de seguridad, buscando mantener la vía aérea permeable.
• Si el niño no respira, pasaremos a las ventilaciones.
6. Ventilación (B):
• Administrar 5 insuflaciones de rescate lentas, de aproximadamente 1 segundo de duración, realizando inspiraciones profundas entre cada respiración para aumentar la cantidad de oxígeno suministrado.
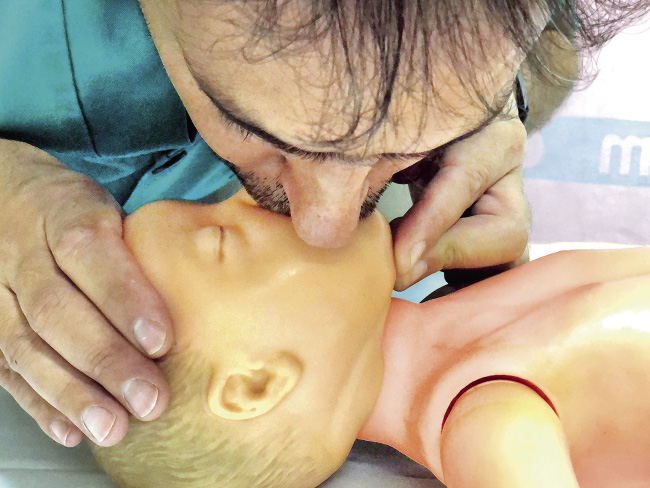
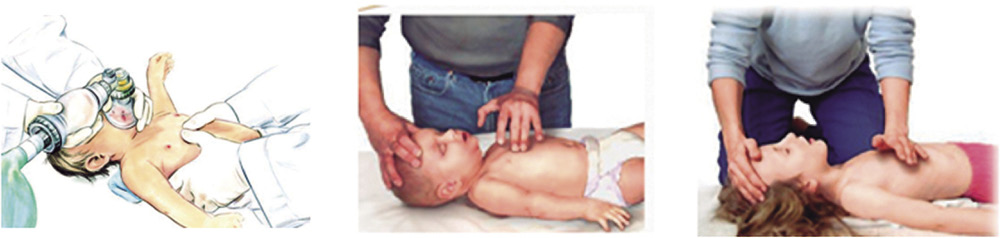
• En los lactantes se utilizará la técnica boca a boca-nariz (Fig. 4). Si el tamaño del lactante no lo permitiese, se podrá intentar sellar solo la boca o la nariz del lactante. Podemos ayudarnos de un paño enrollado debajo de los hombros para mantener la cabeza en posición neutra.

Figura 4. Ventilación boca a boca-nariz.
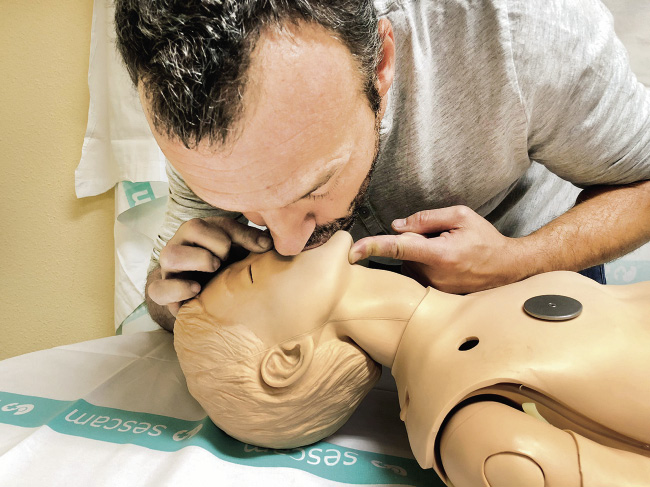
• En los niños se hará boca a boca (Fig. 5). El reanimador debe hacer pinza en la parte blanda de la nariz con los dedos pulgar e índice de la mano que tiene apoyada en su frente, para asegurarse de que el aire de la insuflación de rescate no se fuga por la nariz.
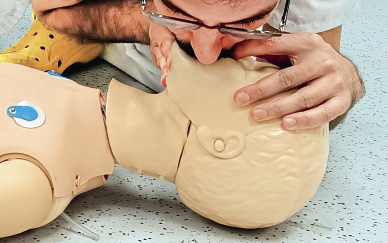
Figura 5. Ventilación boca a boca.
• Se debe comprobar la efectividad de las respiraciones de rescate, observando que producen elevación y descenso del tórax. Al menos, 2 de estas 5 insuflaciones de rescate han de ser efectivas.
• Si no se producen movimientos torácicos durante la aplicación de las respiraciones de rescate, se debe volver a valorar la vía aérea del niño: reposicionar la cabeza, apartar cualquier obstrucción visible y asegurar un buen sellado.
• Si a pesar de estas maniobras no se consigue que el tórax del niño se expanda, se debe considerar la existencia de un cuerpo extraño que obstruye la vía aérea, por lo que no nos demoraremos con las ventilaciones y pasaremos a realizar las compresiones torácicas.
• Cuando hay solo un reanimador y este tiene teléfono móvil, se debe llamar al 112 en modo “manos libres” y continuar con la RCP mientras se espera una respuesta.
7. Valoración de la circulación (C):
• Tras realizar las respiraciones de rescate, habrá que comprobar el estado circulatorio mediante la búsqueda de “signos de vida” (movimientos, tos o respiraciones normales), sin emplear para ello más de 10 segundos.
• En el caso del personal sanitario, se puede palpar el pulso de una arteria central: braquial (o femoral) en el lactante, y carotídeo (o femoral) en el niño.
• Si se detectan signos de vida o si se palpa con seguridad un pulso central a una frecuencia mayor de 60 latidos por minuto, continuar manteniendo la apertura de la vía aérea, administrando entre 12 y 20 ventilaciones por minuto, según la edad del niño. Reevaluar continuamente la situación respiratoria y circulatoria hasta que llegue personal del SEM o más cualificado, o hasta que se recupere la respiración espontánea, debiéndose colocar al niño en posición lateral de seguridad en ese momento.
• Si no hay signos de vida, no existe pulso central o la frecuencia es menor de 60 latidos por minuto, se deben iniciar las compresiones torácicas.
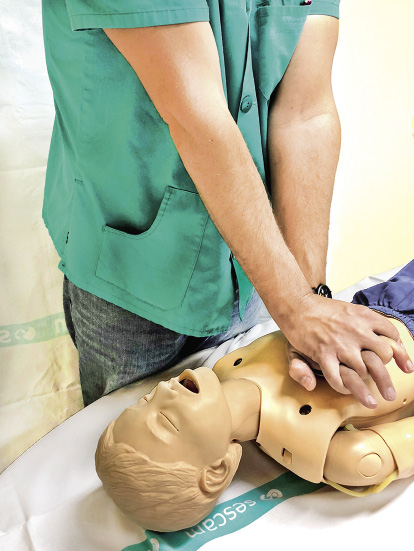
8. Compresiones torácicas (C):
• La compresión debe realizarse en la mitad inferior del esternón, evitando comprimir el apéndice xifoides.
• Se debe comprimir un tercio del diámetro anteroposterior del tórax, que corresponde a unos 4 cm en lactantes y unos 5 cm en niños.
• La frecuencia de las compresiones será 100-120 compresiones por minuto.
• En lactantes, se podrá realizar mediante:
– Técnica con dos dedos (Fig. 6): preferible cuando existe un único reanimador; este colocará las puntas de dos dedos en la mitad inferior del esternón y realizará 15 compresiones, evitando comprimir xifoides, abdomen o costillas. La otra mano podrá mantenerse en la frente para mantener la vía aérea abierta.

Figura 6. Compresiones torácicas con dos dedos.
– Técnica abarcando el tórax con las dos manos (Fig. 7): preferible cuando existen dos o más reanimadores; el reanimador se sitúa a los pies del lactante y coloca ambos pulgares sobre la mitad inferior del esternón, en el mismo punto que con la técnica anterior, mientras abraza el tórax con ambas manos.

Figura 7. Compresiones torácicas abarcando el tórax del lactante.
• En niños, se podrá realizar:
– Colocando el talón de una mano sobre el esternón (Fig. 8).

Figura 8. Compresiones torácicas con una mano.
– Con las dos manos con los dedos entrelazados, apoyando el talón de una de ellas sobre el dorso de la otra (Fig. 9).
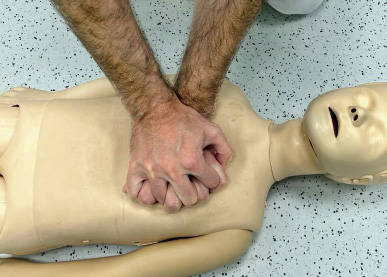
Figura 9. Compresiones torácicas con dos manos.
• Tras cada compresión hay que dejar que el tórax se expanda, es decir, que vuelva a su posición normal, pero evitando separar las manos del pecho del paciente. La compresión debe durar la mitad del ciclo “compresión-descompresión”.
• Relación compresiones torácicas-ventilación:
– Población general: 30 compresiones torácicas-2 ventilaciones.
– Personal sanitario: 15 compresiones torácicas-2 ventilaciones, a no ser que únicamente haya un reanimador. En ese caso podrá utilizar también una relación de 30 compresiones torácicas-2 ventilaciones.
• Para evitar la fatiga, si hay más de un reanimador, estos se irán sustituyendo en el rol de administrar las compresiones. Si hay solo un reanimador, este podrá cambiar la mano que comprime o intercambiar la técnica (de una a dos manos).
9. No se interrumpirá la RCP a menos que haya claros signos de circulación (movimientos, tos) o cuando se esté exhausto.
Desfibrilador externo semiautomático (DESA)
Los DESA son dispositivos compactos que, una vez conectados a los electrodos colocados en el tórax del paciente, tienen la capacidad de analizar el ritmo cardiaco y determinar si es necesario aplicar una descarga eléctrica.
Cuando se detecta un ritmo cardiaco susceptible de ser desfibrilado, el dispositivo emite una señal visual y auditiva, señalando la presencia de un ritmo desfibrilable, y se carga automáticamente con una cantidad predeterminada de energía. Esta energía normalmente es de 150-360 julios, aunque algunos equipos permiten atenuar la dosis de energía, sobre 50-75 julios. Si el equipo dispone de atenuadores, los deberemos utilizar cuando el paciente tenga una edad inferior a 8 años; si no dispusiese de ellos, se recomienda utilizar el DESA de adulto de todas formas (es más adecuado administrar alta energía que no administrar ninguna descarga a un niño en situación de PCR y ritmo desfibrilable). La descarga solo se aplicará sobre el paciente si el operador presiona el botón correspondiente.
Deberemos utilizar el DESA durante la RCP pediátrica de origen cardiaco y con posible ritmo desfibrilable. Este origen lo sospecharemos por un colapso súbito presenciado sin signos de etiología respiratoria o traumática. La integración del DESA en la RCP, se realiza de la siguiente manera:
1. En el caso de existir un reanimador, este realizará un minuto de RCP antes de ir a buscar el DESA. Con dos o más reanimadores, uno realizará la RCP, mientras que el otro pedirá ayuda y buscará el DESA más cercano.
2. Una vez que dispongamos del DESA, colocaremos los parches en el paciente, sin interrumpir las maniobras de RCP en el caso de ser dos o más reanimadores.
En los niños mayores de 8 años colocaremos uno de los parches a la derecha del esternón debajo de la clavícula, y el otro de manera longitudinal, paralelo al anterior, en el tórax en la línea medioaxilar izquierda (Fig. 10).

Figura 10. Colocación de los parches del DESA (desfibrilador externo semiautomático) en niños mayores de 8 años.
En los niños menores de 8 años se colocarán de la misma manera, salvo cuando se tuviesen que utilizar parches de adultos. En ese caso, estos se colocarán uno en el esternón y otro en la espalda.
3. Encenderemos el DESA y seguiremos sus indicaciones:
• Si el DESA indica la administración de una descarga, deberemos asegurarnos de que nadie toque a la víctima y entonces pulsar el botón de descarga. Después de esto continuaremos con las maniobras de RCP. A los 2 minutos, el DESA se activará automáticamente y analizará el ritmo cardiaco de nuevo.
• Si el dispositivo no indica dar una descarga, continuaremos con las maniobras de RCP.
4. No se deben retirar los parches ni apagar el DESA hasta que no llegue el SEM.
Obstrucción de la vía aérea por un cuerpo extraño (OVACE)
Deberemos sospechar una OVACE ante un episodio de dificultad respiratoria que comienza: de forma brusca, asociado a tos, náuseas o estridor, que se produce durante la comida o durante el juego con objetos pequeños y sin otros signos de enfermedad.
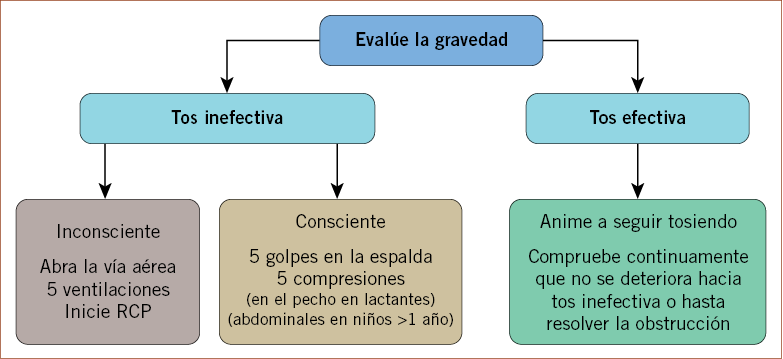
El tratamiento de la OVACE será el siguiente:
1. Si el niño tose de manera efectiva: se tranquilizará al niño, se le animará a toser y se le vigilará continuamente hasta la expulsión del cuerpo extraño.
2. Si el niño tose de manera inefectiva, la tos anterior se transforma en inefectiva o el niño no tose en absoluto: se activará el SEM.
En el caso de que el niño presente tos ineficaz y esté consciente, deberemos realizar las siguientes maniobras con el objetivo de desplazar el cuerpo extraño hasta conseguir una respiración adecuada:
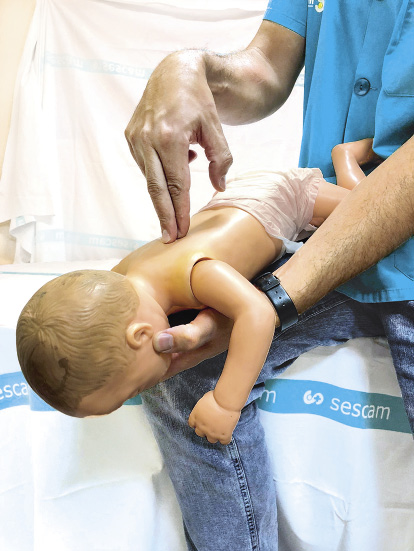
• Si se trata de un lactante:
– Sentarse o arrodillarse para poder sujetar al lactante de manera segura. Colocar al lactante boca abajo sobre el antebrazo, apoyándose sobre su muslo, sujetando la cabeza del lactante, poniendo el pulgar de la mano en un ángulo de la mandíbula y uno o dos dedos de la misma mano en el ángulo contrario de la mandíbula. La cabeza del lactante quedará en una posición inferior respecto del resto de su cuerpo.
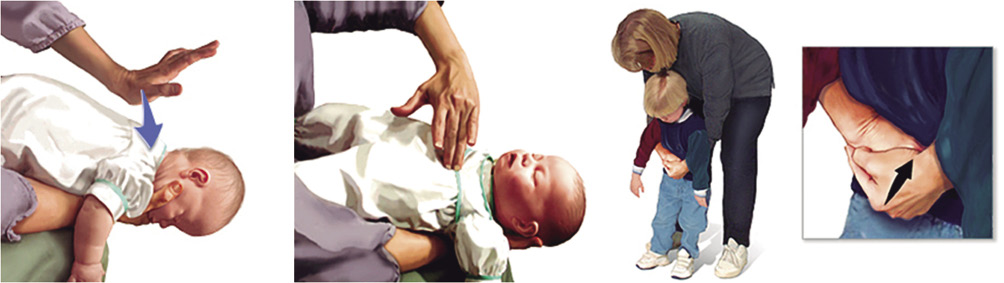
– Realizar 5 golpes secos con el talón de la otra mano en la región interescapular de la espalda (Fig. 11).

Figura 11. Golpes interescapulares en lactante con tos no efectiva.
– Voltear al lactante, pasándolo al otro antebrazo y poniéndolo en posición supina, manteniendo su cabeza sujeta con la mano en una posición inferior. Observar si el cuerpo extraño se ha movilizado hasta la boca y puede extraerse con seguridad. Si no es así, realizaremos 5 compresiones torácicas (Fig. 12).
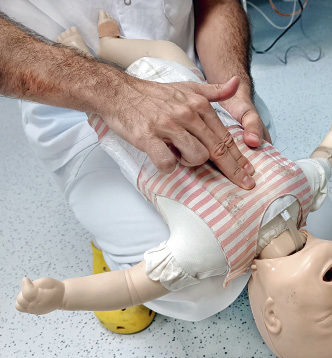
Figura 12. Compresiones torácica en lactante con tos no efectiva.
– Realizar las 5 compresiones torácicas con el dedo índice y pulgar en la mitad inferior del esternón hacia la espalda y la cabeza, de manera similar a las compresiones torácicas de la RCP, pero más secas y dadas con una frecuencia menor.
• Si se trata de un niño mayor de un año:
– Colocar al niño incorporado, pero ligeramente hacia delante, con la cabeza en una posición más baja que el resto del cuerpo.
– Dar 5 golpes en la región interescapular de la espalda (Fig. 13).

Figura 13. Golpes interescapulares en niño con tos no efectiva.
– Si el objeto no ha sido expulsado, nos situaremos por detrás del niño, pasando nuestros brazos por debajo de sus axilas y abrazando su torso. Cerrar el puño de la mano dominante y situarlo en el epigastrio. Sujetar el puño con la otra mano y comprimir 5 veces hacia adentro y hacia arriba (maniobra de Heimlich). Asegurarse de que la presión no se aplica sobre la apófisis xifoides ni sobre las costillas (Fig. 14).
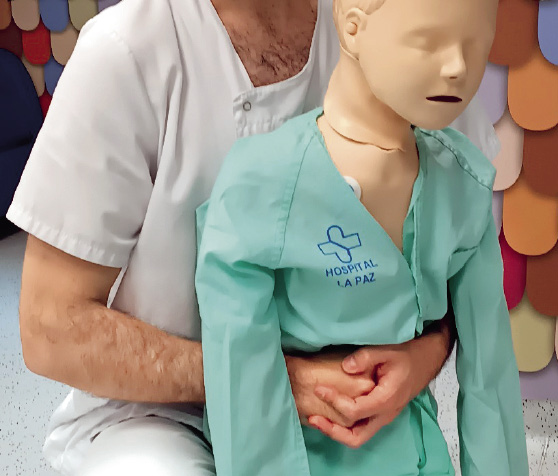
Figura 14. Maniobra de Heimlich en niño con tos no efectiva.
En el caso de que el niño presente tos ineficaz y esté inconsciente deberemos:
• Colocar al niño en una superficie plana y firme.
• Solicitar ayuda de la misma manera descrita en la RCP básica, según haya uno o más reanimadores.
• Abrir la vía aérea y buscar algún objeto visible. Si se ve, intentaremos extraerlo de manera segura mediante un barrido con un dedo. Nunca se deberá realizar un barrido a ciegas, por la posibilidad de impactar el cuerpo extraño más profundamente.
• Administrar 5 ventilaciones de rescate, valorando su eficacia.
• Si tras administrar las 5 ventilaciones de rescate no hay signos de vida, iniciar las compresiones torácicas.
• Revisar la cavidad oral durante las respiraciones, para comprobar si hay algún cuerpo extraño e intentar extraerlo de manera segura mediante la técnica de barrido con un dedo.
Cambios respecto a las recomendaciones de 2015
Las recomendaciones europeas de RCP básica en niños mantienen el orden ABC (vía aérea, respiración y compresiones torácicas) en la secuencia de las maniobras. Los cambios fundamentales son:
• Activación del sistema de emergencias. Tras las 5 insuflaciones de rescate, si solo hay un reanimador y dispone de teléfono con llamada con manos libres, se recomienda poner el teléfono móvil en altavoz y llamar para pedir ayuda al SEM mientras se continúa con la RCP. Si no tiene teléfono móvil, se realizará la RCP durante un minuto y, en ese momento, se activará el aviso al SEM.
• Solamente en el caso en que el reanimador observe que el niño presenta una pérdida brusca de consciencia y sospeche que es de origen cardiaco, debe llamar primero al SEM y, a continuación, empezar la reanimación, porque puede que el niño necesite una desfibrilación. Si hay más de un reanimador, uno de ellos debe iniciar inmediatamente la RCP, mientras que el otro busca la ayuda.
• Valoración de la ventilación. Se sugiere que la población general pueda valorar la existencia de respiración solo por la existencia o no de movimientos respiratorios, poniendo una mano sobre el tórax o el abdomen para comprobar si estos se mueven.
• Comprobación de la eficacia de la RCP. Para disminuir al máximo el tiempo sin reanimación, se sugiere no interrumpir la RCP salvo que existan signos claros de circulación (movimientos, tos).
Conflicto de intereses
No hay conflicto de interés en la elaboración del manuscrito. Declaración de intereses: ninguno.
Bibliografía
1. López-Herce J, García C, Domínguez P, Rodríguez-Núñez A, Carrillo Á, Calvo C, et al. Outcome of out-of-hospital cardiorespiratory arrest in children. Pediatr Emerg Care. 2005; 21: 807-15.
2. Meyer L, Stubbs B, Fahrenbruch C, Maeda Ch, Harmon K, Eisenberg M, et al. Incidence, causes, and survival trends from cardiovascular-related sudden cardiac arrest in children and young adults 0 to 35 years of age: A 30-year review. Circulation. 2012; 126: 1363-72.
3. Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, et al. Heart Disease and Stroke Statistics – 2020 Update a Report from the American Heart Association. 2020; 141: e139-e596.
4. Van de Voorde P, Turner NM, Djakow J, de Lucas N, Martinez-Mejias A, Biarent D, et al. European Resuscitation Council Guidelines 2021: Paediatric Life Support. Resuscitation. 2021; 161: 327-87.
5. Bagnall RD, Weintraub RG, Ingles J, Duflou J, Yeates L, Lam L, et al. A Prospective Study of Sudden Cardiac Death among Children and Young Adults. N Engl J Med. 2016; 374: 2441-52.
6. Nolan JP, Maconochie I, Soar J, Olasveengen TM, Greif R, Wyckoff MH, et al. Executive Summary: 2020 International Consensus on Cardiopulmonary Resuscitation and Emergency Cardiovascular Care Science With Treatment Recommendations. Circulation. 2020; 142: S2-S27.
7. Berg KM, Bray JE, Ng K-C, Liley HG, Greif R, Carlson JN, et al. 2023 International Consensus on Cardiopulmonary Resuscitation and Emergency Cardiovascular Care Science With Treatment Recommendations: Summary From the Basic Life Support; Advanced Life Support; Pediatric Life Support; Neonatal Life Support; Education, Circulation. 2023; 148: e187-e280.
8. Maconochie IK, Bingham R, Eich C, López-Herce J, Rodríguez-Núñez A, Rajka T, et al. European Resuscitation Council Guidelines for Resuscitation 2015. Section 6. Paediatric life support. Resuscitation. 2015; 95: 223-48.









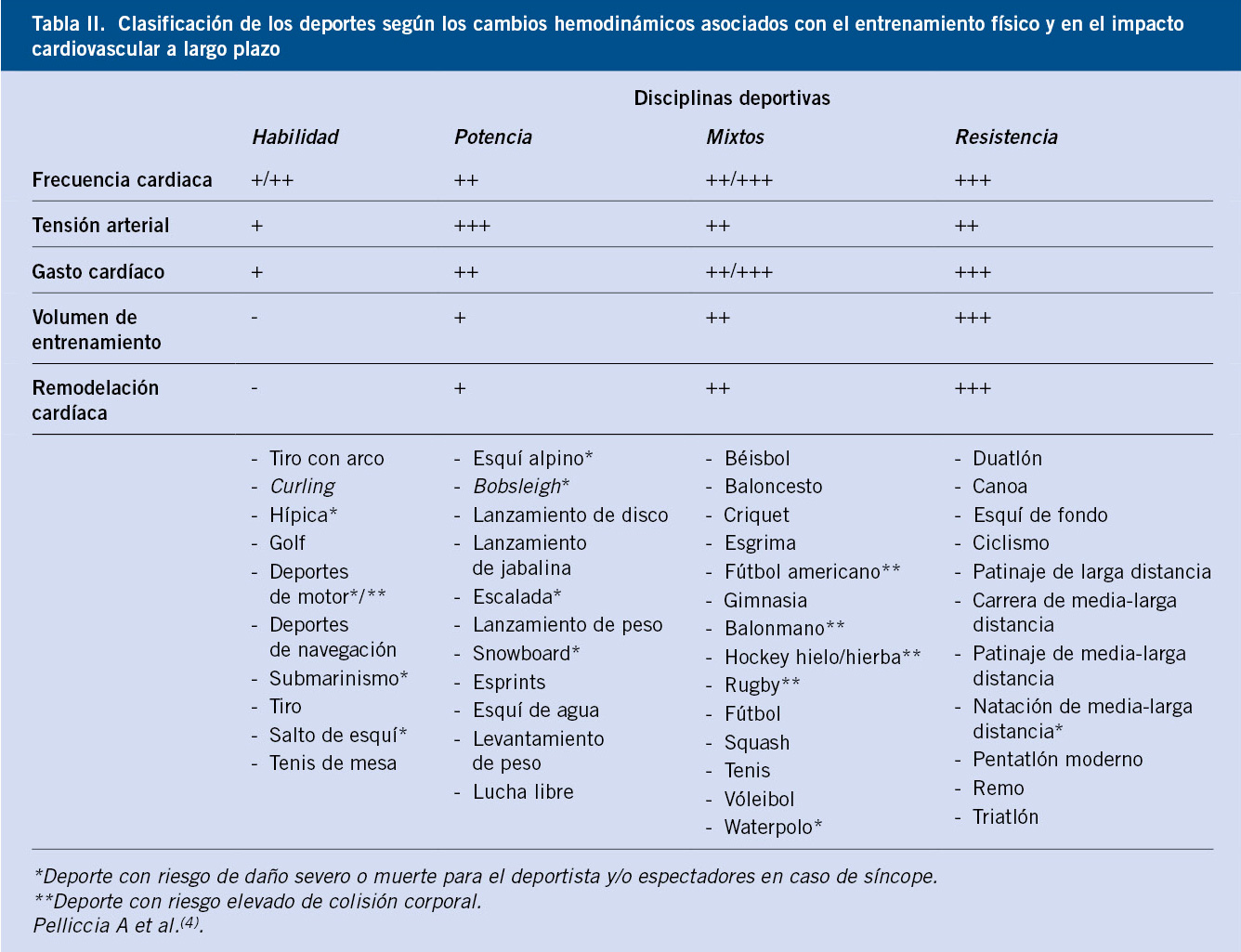
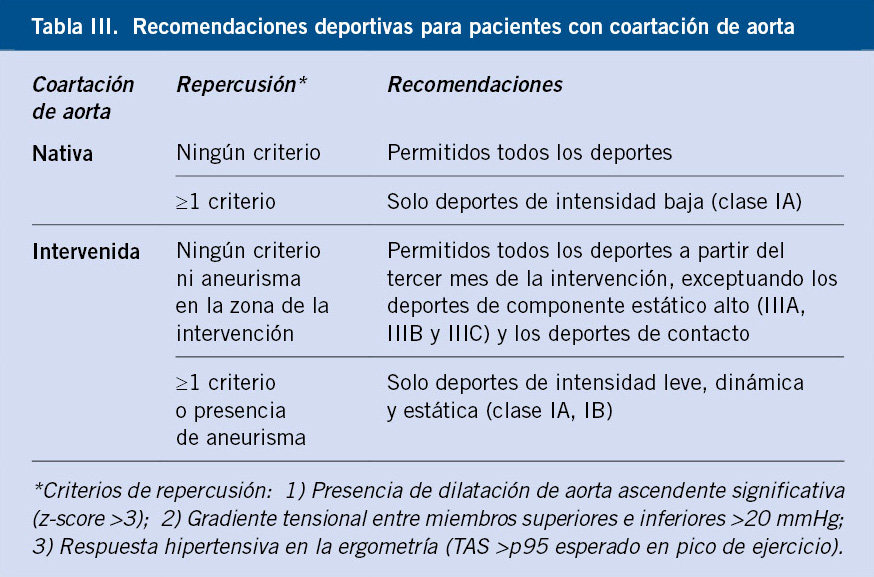


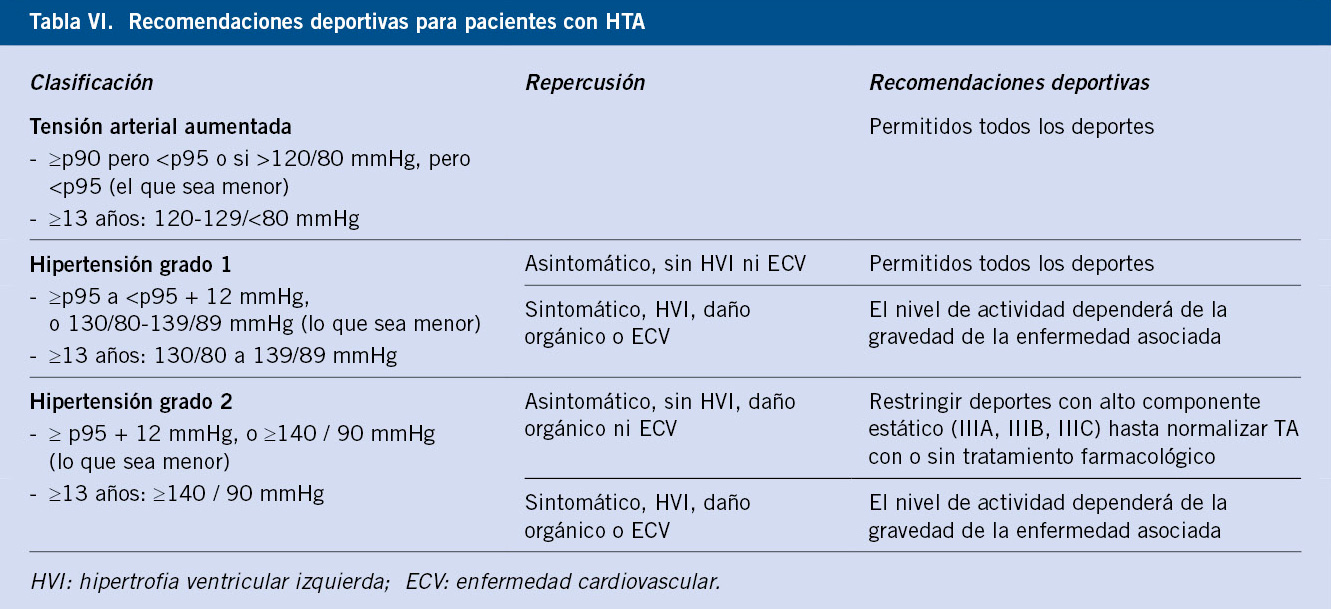


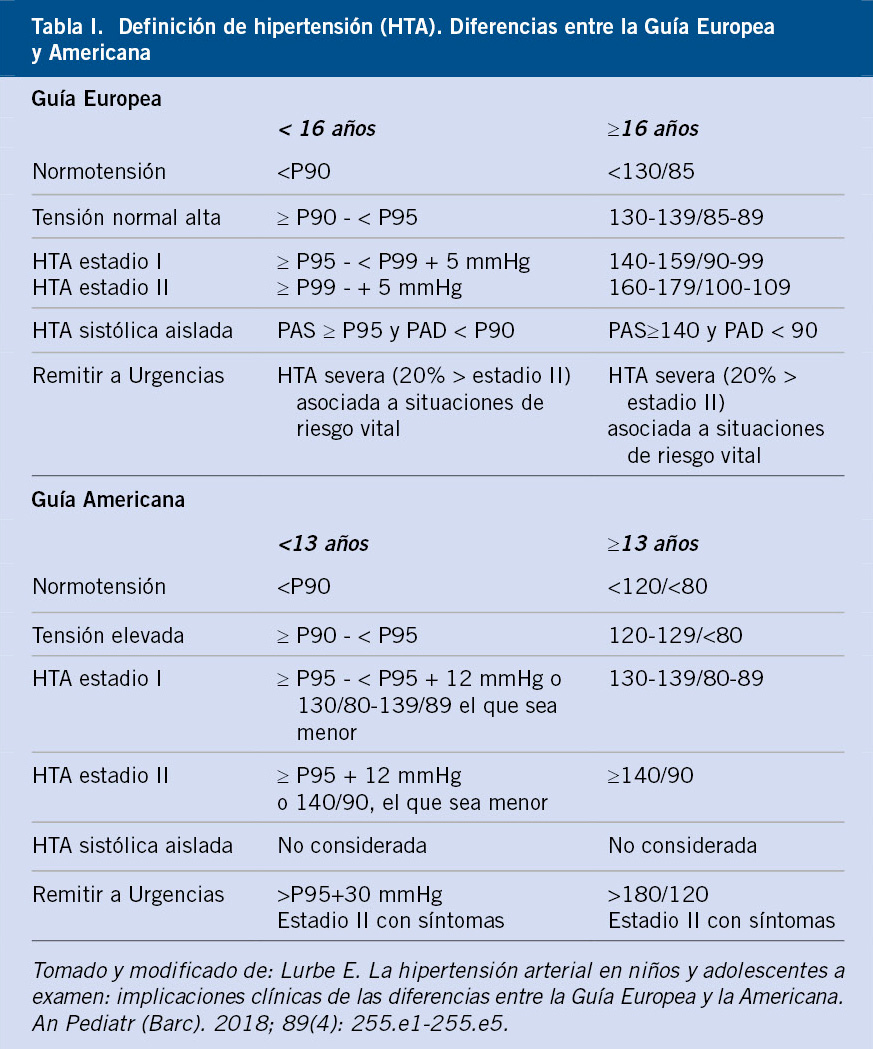


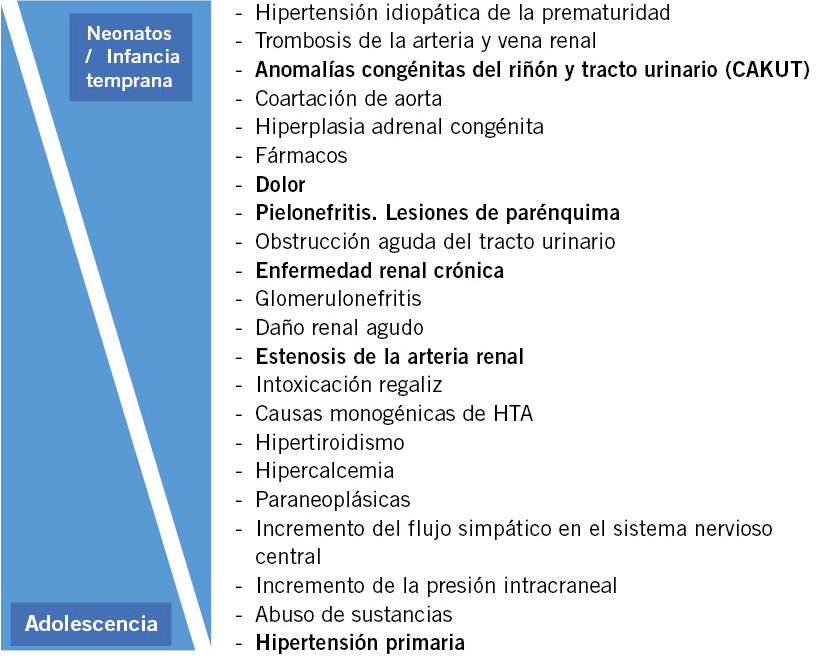
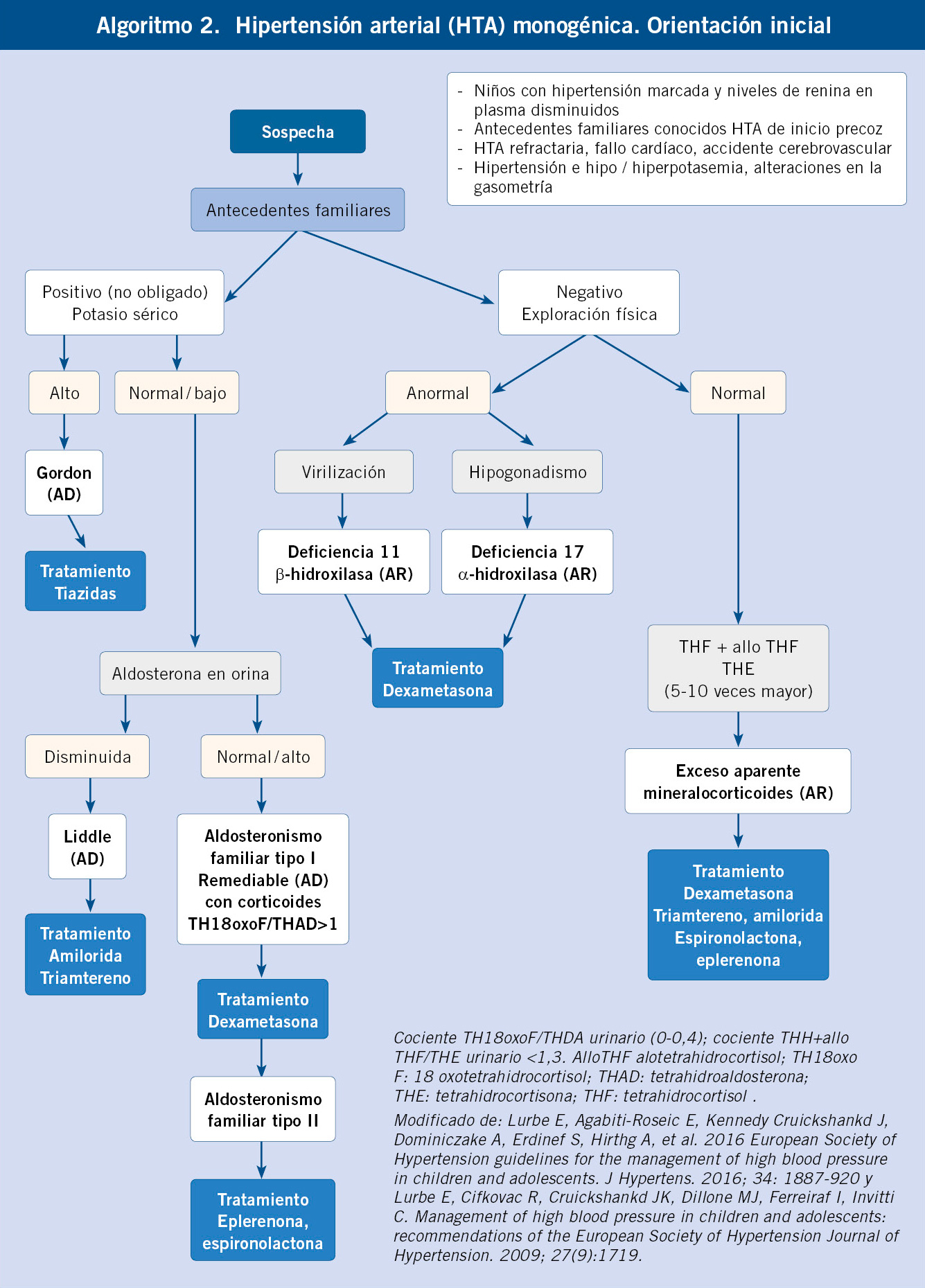



















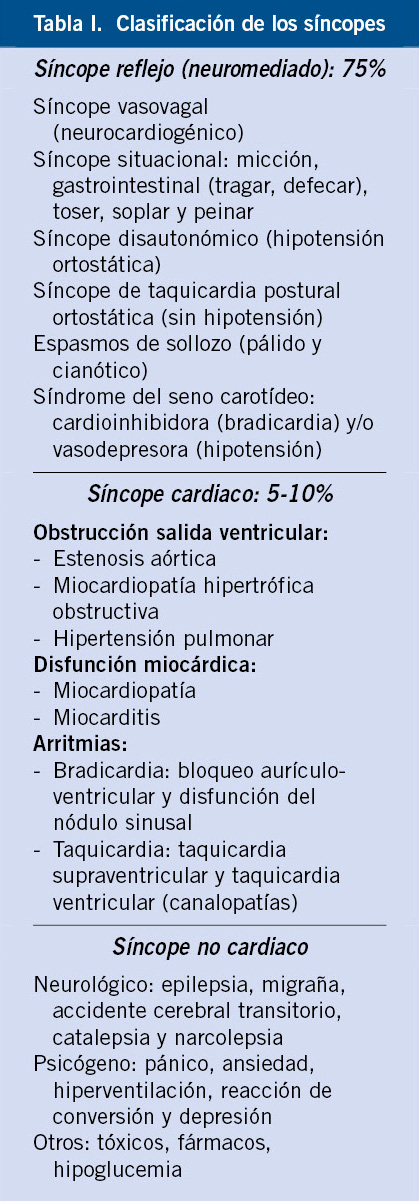




 En Pediatría existe escasa evidencia científica acerca de los factores de riesgo cardiovascular (FRCV). Sin embargo, extrapolando los datos obtenidos de pacientes adultos, es razonable asumir que un correcto abordaje de los FRCV disminuirá la aparición de enfermedad cardiovascular(1).
En Pediatría existe escasa evidencia científica acerca de los factores de riesgo cardiovascular (FRCV). Sin embargo, extrapolando los datos obtenidos de pacientes adultos, es razonable asumir que un correcto abordaje de los FRCV disminuirá la aparición de enfermedad cardiovascular(1). El manejo de los FRCV en Pediatría, consiste en comprobar la adherencia a un estilo de vida saludable (prevención primordial) e identificar y tratar a los niños en riesgo de arterioesclerosis temprana (prevención primaria y secundaria)(2).
El manejo de los FRCV en Pediatría, consiste en comprobar la adherencia a un estilo de vida saludable (prevención primordial) e identificar y tratar a los niños en riesgo de arterioesclerosis temprana (prevención primaria y secundaria)(2).  En las revisiones de salud, se debe obtener información y dar pautas sobre: dieta, actividad física y exposición al tabaco. Se revisará el patrón de sueño y se identificarán los antecedentes familiares de enfermedad cardiovascular prematura(1,2).
En las revisiones de salud, se debe obtener información y dar pautas sobre: dieta, actividad física y exposición al tabaco. Se revisará el patrón de sueño y se identificarán los antecedentes familiares de enfermedad cardiovascular prematura(1,2).  Se recomienda el cribado universal de obesidad mediante la monitorización de la talla, el peso y el índice de masa corporal (IMC) en las revisiones de salud, a partir de los 6 años(3). USPSTF* 2017, grado de recomendación B.
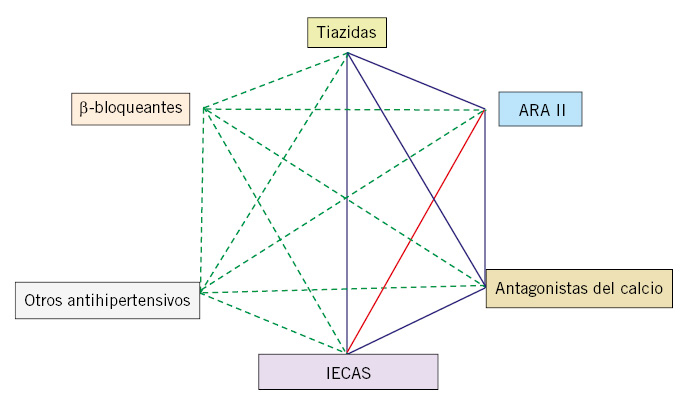
Se recomienda el cribado universal de obesidad mediante la monitorización de la talla, el peso y el índice de masa corporal (IMC) en las revisiones de salud, a partir de los 6 años(3). USPSTF* 2017, grado de recomendación B.  Se recomienda el cribado universal de hipertensión arterial (HTA), mediante la monitorización de la tensión arterial en las revisiones
Se recomienda el cribado universal de hipertensión arterial (HTA), mediante la monitorización de la tensión arterial en las revisiones No se recomienda un cribado universal de diabetes mellitus (DM)(5). ADA 2018, grado de recomendación A.
No se recomienda un cribado universal de diabetes mellitus (DM)(5). ADA 2018, grado de recomendación A.  Se realizará cribado de diabetes mellitus tipo 2 (DM2) mediante la determinación de glucemia en ayunas cada 2 años, en aquellos pacientes mayores de 10 años que presenten sobrepeso (IMC > percentil 85 para la edad y sexo) asociado a 2 o más de los siguientes factores de riesgo: DM2 en familiar de primer o segundo grado, grupo étnico de riesgo, signos de resistencia insulínica (acantosis, dislipemia, HTA o síndrome de ovario. poliquístico)(5). ADA 2018, grado de recomendación A
Se realizará cribado de diabetes mellitus tipo 2 (DM2) mediante la determinación de glucemia en ayunas cada 2 años, en aquellos pacientes mayores de 10 años que presenten sobrepeso (IMC > percentil 85 para la edad y sexo) asociado a 2 o más de los siguientes factores de riesgo: DM2 en familiar de primer o segundo grado, grupo étnico de riesgo, signos de resistencia insulínica (acantosis, dislipemia, HTA o síndrome de ovario. poliquístico)(5). ADA 2018, grado de recomendación A  La recomendación de un cribado universal de dislipemia (DL), mediante un perfil lipídico (colesterol total, triglicéridos y lipoproteínas) a los 9-11 años y a los 17-20 años es controvertida en el momento actual(1,6). USPSTF* 2016, grado recomendación I.
La recomendación de un cribado universal de dislipemia (DL), mediante un perfil lipídico (colesterol total, triglicéridos y lipoproteínas) a los 9-11 años y a los 17-20 años es controvertida en el momento actual(1,6). USPSTF* 2016, grado recomendación I.  Se recomienda el cribado de DL con perfil lipídico en mayores de 2 años, cada 3-5 años si existen los siguientes factores de riesgo: enfermedad cardiovascular prematura en la familia, hipercolesterolemia familiar, comorbilidades (HTA, DM, obesidad)(1,6). AHA 2019, grado de recomendación B.
Se recomienda el cribado de DL con perfil lipídico en mayores de 2 años, cada 3-5 años si existen los siguientes factores de riesgo: enfermedad cardiovascular prematura en la familia, hipercolesterolemia familiar, comorbilidades (HTA, DM, obesidad)(1,6). AHA 2019, grado de recomendación B. Se individualizará el seguimiento de los FRCV clásicos en presencia de otras patologías de alto riesgo (enfermedad renal crónica, enfermedades inflamatorias, cáncer y cardiopatías congénitas)(1).
Se individualizará el seguimiento de los FRCV clásicos en presencia de otras patologías de alto riesgo (enfermedad renal crónica, enfermedades inflamatorias, cáncer y cardiopatías congénitas)(1).