|
| Temas de FC |
S. Barrena Delfa*, A.L. Luis Huertas**
*Servicio de Cirugía Pediátrica. Hospital Universitario La Paz. Madrid. **Servicio de Cirugía Pediátrica. Hospital Universitario Niño Jesús. Madrid
| Resumen
Las malformaciones congénitas del tracto digestivo constituyen un espectro variado de patologías, que incluyen: la aparición de atresias o estenosis del tubo digestivo, duplicaciones del tracto intestinal, mal posicionamiento de estructuras o deformidades anatómicas que pueden provocar alteraciones, tanto en el periodo neonatal como a lo largo de la infancia.
|
| Abstract
Congenital malformations of the gastrointestinal tract constitute a wide spectrum of pathologies that include the appearance of atresias or stenosis of the gastrointestinal tract, duplications of the intestine, poor positioning of structures or anatomical deformities that can cause alterations both in the neonatal period and throughout childhood. |
Palabras clave: Atresia esofágica; Atresia intestinal; Atresia anal; Atresia duodenal; Malrotación intestinal.
Key words: Esophageal atresia; Intestinal atresia; Anal atresia; Duodenal atresia; Intestinal malrotation.
Pediatr Integral 2019; XXIII (6): 301 – 309
Malformaciones congénitas digestivas
Diagnóstico prenatal de las malformaciones digestivas
El diagnóstico prenatal de las malformaciones digestivas cada vez es más acertado, gracias a herramientas de control ecográfico prenatal y resonancia magnética fetal.
Actualmente, el control del embarazo es realizado en la mayoría de los países desarrollados, como el nuestro, siendo habituales la ecografía prenatal tanto en el primer como en el segundo trimestre; estas dos ecografías diagnósticas permiten detectar la mayoría de las anomalías fetales. En el caso del tracto gastrointestinal, el diagnóstico es más limitado. Como consecuencia de esto, la mayoría de las obstrucciones intestinales no son identificadas hasta el final de la gestación o incluso después el nacimiento.
En aquellos casos en los que existan dudas diagnósticas o que sea necesaria una mayor precisión, existen pruebas que la aportan, como los test genéticos o la resonancia magnética fetal.
Existen peculiaridades diagnósticas prenatales, dependiendo del tipo de patología existente:
• Atresia de esófago: el diagnóstico prenatal de la atresia esofágica es aproximadamente de un 30% según las series, ya que el esófago es un órgano difícil de visualizar prenatalmente. Se puede sospechar mediante signos indirectos como el polihidramnios, que aparece en aproximadamente un 50% de los casos(1). Otros signos, como la ausencia de cámara gástrica, parecen estar más relacionados con la atresia sin fístula.
• Atresia duodenal: su diagnóstico suele ser más preciso y se llega a él en un 75% de los casos. El signo prenatal clásico es la “doble burbuja”, que consiste en la dilatación de la cámara gástrica y el duodeno proximal dilatado. Habitualmente, también se asocia a polihidramnios. Un tercio de ellos se asocia con trisomía 21.
• Obstrucción intestinal: algunas malformaciones como las atresias intestinales distales debutan con signos de obstrucción intestinal prenatal. Los hallazgos son asas distendidas y peristálticas, acompañados de polihidramnios. No siempre la presencia de asas dilatadas se corresponde con malformaciones congénitas, en ocasiones, es un signo de otras enfermedades como la fibrosis quística(2).
• Quistes de duplicación intestinal: este tipo de malformaciones congénitas son raras y pueden aparecer a lo largo del tracto gastrointestinal. Son estructuras tubulares o quísticas de tamaño variable, ocasionalmente aisladas o asociadas con otras malformaciones gastrointestinales. Es importante, en estos casos, que se realice un diagnóstico diferencial con otras patologías como: los quistes broncógenos, las malformaciones congénitas de vía aérea u otros quistes localizados en el abdomen(3).
• Malformación anorrectal: el ano y el recto son estructuras difícilmente visibles en la ecografía fetal; de ahí que el diagnóstico de las malformaciones anorrectales sea difícil. Sí que se puede diagnosticar la atresia rectal, en aquellos casos en los que se ve dilatación del recto y se visualiza ecográficamente el complejo anorrectal y el esfínter rectal por debajo de la mucosa del recto.
En los casos en los que se llegue a un diagnóstico prenatal de malformaciones digestivas, es recomendable que la familia del niño sea remitida a un panel de expertos, que incluya tanto especialistas de Pediatría como cirujanos pediátricos, para evaluar el pronóstico de la enfermedad y se pueda discutir la estrategia a seguir en el periodo pre y perinatal.
Atresia de esófago y fístula traqueoesofágica
La atresia de esófago (AE) consiste en la falta de continuidad del esófago, asociado o no a fístula traqueoesofágica (FTE).
Esta patología ocurre en 1/3.000 RNV, y es un reto para el cirujano pediátrico. Se han descrito los factores de riesgo y predictores de la mortalidad relacionados con el peso al nacimiento, las anomalías cromosómicas, renales y cardiacas.
Se han postulado diferentes clasificaciones, pero en general, se dividen en cinco tipos (Fig. 1), siendo la más frecuente aquella que presenta bolsón ciego proximal y FTE distal.
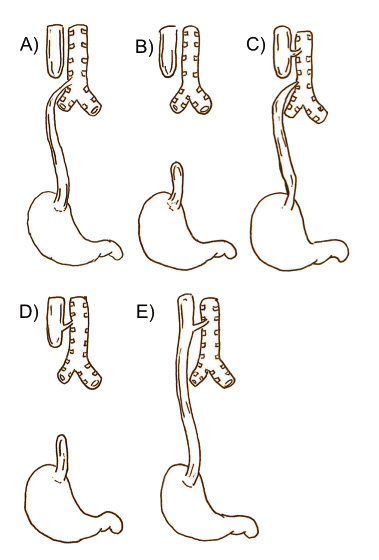
Figura 1. Tipos de atresia de esófago. A. La anomalía más común (86%). B. Atresia pura (8%), generalmente es tipo “long gap”. C. Atresia con fístula proximal y distal (1%). D. Atresia con fístula proximal sin fístula distal (1%), también suele ser tipo “long-gap”. E. Fístula traqueoesofágica sin fístula (4%).
Existe otro tipo en el que existe FTE sin AE, que es conocida como fístula en H. Aquellas en las que el esófago está ciego en ambos lados o que tienen FTE proximal suelen ser atresias con larga distancia entre los cabos (conocidas como atresias “long-gap”), y estas son más difíciles de reparar en el periodo neonatal inmediato(4).
La AE, por otro lado, puede aparecer aislada, pero, en ocasiones, se acompaña de otras condiciones como la prematuridad, y dos tercios de los casos presentan otras anomalías, que bien son cromosómicas o relacionadas con la asociación VACTERL (vertebrales, anorrectales, cardiacas, traqueales, esofágicas, renales y de miembros).
Al nacimiento, los pacientes con AE pueden presentar dificultad para tragar, episodios de tos o cianosis, ocasionalmente, al intentar la primera toma. Existe impedimento para pasar una sonda esofágica y, al realizar una radiografía de tórax, se ve como esta hace un bucle en el bolsón superior (Fig. 2).

Figura 2. Radiografía de tórax de un paciente con atresia de esófago, en la que se visualiza sonda nasogástrica con un bucle en el bolsón superior esofágico.
La presencia de gas abdominal nos hace sospechar la presencia de una FTE y, la ausencia de gas, una atresia intestinal “pura”. La asociación VACTERL se pone de manifiesto con el examen físico y radiológico; de ahí, que sea importante revisar los miembros y el periné del niño, así como realizar radiografías de tórax y abdomen, ecografía y ecocardiograma.
Al nacimiento, es conveniente poner al paciente con la cabeza elevada unos 30-45º y con una sonda de doble luz con aspiración continua para prevenir la aspiración. El tratamiento definitivo consiste en poner en continuidad el esófago y en el cierre de la FTE. La corrección se puede realizar en una sola intervención, que consiste en realizar una broncoscopia para localizar la fístula, disecar la FTE, y tras seccionar y cerrar la fístula, realizar una anastomosis término-terminal esofágica. Habitualmente, se deja una sonda nasogástrica (SNG) como tutor de la anastomosis, por la que se puede alimentar tras la intervención (Fig. 3). En ocasiones, es necesario colocar un tubo de tórax para drenaje.
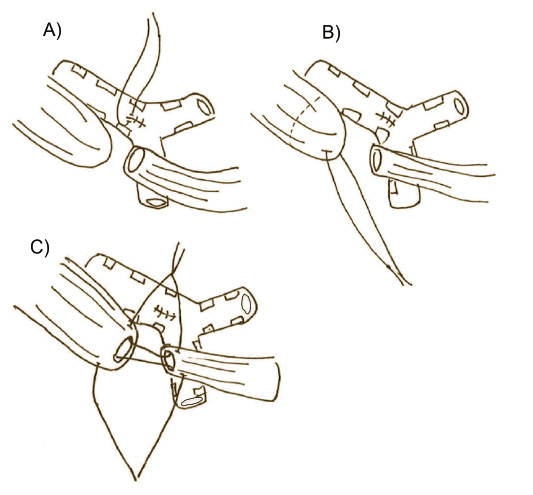
Figura 3. Esquema de los pasos de la intervención de la atresia esofágica. A. Sección de la fístula traqueoesofágica y cierre de la misma. B. Disección del bolsón proximal. C. Anastomosis término-terminal del esófago.
La alimentación se puede iniciar por la SNG a partir del 2º-3er día. Se suele realizar un esofagograma al 5º-7º día, para comprobar la integridad de la anastomosis. Si esta es adecuada, proceder a la alimentación oral y a la retirada del tubo de tórax.
La atresia tipo long-gap suele requerir otras técnicas como: la elongación esofágica, la anastomosis primaria diferida o la sustitución esofágica, lo cual depende de la experiencia de su cirujano responsable(4).
Las complicaciones más habituales de la AE son: la fuga de la anastomosis, la recurrencia de la FTE y la estenosis de la unión esofágica. Los pacientes con AE tienen más tendencia al reflujo gastroesofágico y a la traqueomalacia(5).
La mayoría de los niños con AE presentan un buen pronóstico a largo plazo, con una alimentación normal, así como su crecimiento y desarrollo, aunque en estos pacientes existen problemas de deglución, así como de peristalsis esofágica(6,7).
Atresia y estenosis duodenal
La atresia o estenosis duodenal consiste en la obstrucción completa o parcial del duodeno que puede ser debida: a una atresia, a la presencia de una membrana o a un páncreas anular.
El diagnóstico prenatal cada vez se realiza con más frecuencia y el signo clásico es la presencia de doble burbuja en la radiografía abdominal al nacimiento (Fig. 4).

Figura 4. A. Radiografía de tórax-abdomen, en la que se visualiza una imagen de doble burbuja. A la derecha, se muestra un esquema de los diferentes tipos de atresia duodenal. B. Páncreas anular. C. Atresia duodenal completa. D. Atresia duodenal con membrana.
El diagnóstico diferencial se debe hacer con malrotación y vólvulo intestinal. Un 20% de los niños asocia malrotación y un 30% síndrome de Down.
La clínica suele consistir en: vómitos, generalmente biliosos y distensión epigástrica. El diagnóstico se realiza con radiografía simple de abdomen y, si hubiera paso de gas distal, se debería pensar que es una estenosis duodenal.
El tratamiento es quirúrgico. Antes de la intervención es importante colocar una sonda nasogástrica para descomprimir el estómago. La intervención consiste en realizar una duodeno-duodenostomía en diamante, en el caso de las atresias y el páncreas anular; en el caso de las estenosis duodenales, el tratamiento consiste en la apertura de la membrana. Este procedimiento se puede realizar por laparotomía o por laparoscopia, dependiendo de la experiencia del cirujano(8). El manejo postoperatorio consiste en antibioterapia, nutrición parenteral y descompresión gástrica con sonda hasta el inicio de la tolerancia oral. En ocasiones, la nutrición enteral completa tarda en conseguirse, debido a la mala motilidad gástrica y duodenal; de ahí, que algunos pacientes presenten ingresos prolongados.
Atresia intestinal
La atresia intestinal consiste en una ausencia de continuidad de la luz intestinal, que puede aparecer en cualquier punto del intestino delgado y grueso.
Es una de las causas de obstrucción intestinal en el neonato. Se pueden localizar en cualquier punto del intestino delgado y grueso, aunque la localización yeyuno-ileal es la más frecuente. La etiopatogenia tiene varias teorías, aunque la más aceptada es la teoría del proceso isquémico intestinal, que provoca una necrosis y cicatrización del intestino.
Las atresias yeyuno ileales suelen ser solitarias, aunque existen diferentes tipos. En ocasiones, se presentan como poliatresias o como atresias tipo “Apple peel”, en las que se asocia un defecto de meso y un íleon corto (Fig. 5).

Figura 5. A. Imagen quirúrgica de una atresia intestinal tipo 1. B. Imagen quirúrgica de una atresia intestinal tipo “Apple peel” (3b). C. Esquema que muestra la clasificación de las atresias intestinales.
Aquellas que presentan formas complejas, pueden presentar a medio plazo un intestino corto, debido a la ausencia de longitud intestinal y a las múltiples cirugías que requieren(9).
Prenatalmente, se pueden sospechar por presentar: una importante distensión de asas, la presencia de polihidramnios y retraso de crecimiento intrauterino. Asocian con cierta frecuencia prematuridad y bajo peso y, al nacimiento presentan: una importante distensión abdominal, con peristaltismo marcado y con vómitos biliosos, al principio, ocasionales y después continuos.
El diagnóstico se realiza mediante radiografía simple de abdomen, visualizándose una obstrucción intestinal con niveles hidroaéreos en las asas. Ocasionalmente, presentan calcificaciones, imagen que nos puede orientar hacia un proceso de peritonitis meconial.
El tratamiento consiste en la descompresión gástrica, la fluidoterapia y la antibioterapia hasta la intervención quirúrgica, la cual consiste en poner en continuidad el tubo digestivo, para ello se realiza la resección del segmento atrésico y la anastomosis. Si existe desproporción entre los cabos, hay que realizar un “tapering”, que consiste en adaptar el tamaño de ambos cabos intestinales. En ocasiones, se precisan múltiples resecciones y, en los casos más graves, se puede requerir una yeyunostomía o ileostomía(10).
La atresia cólica es la forma menos frecuente. Tanto la clínica como el diagnóstico y tratamiento son similares a los anteriores y se puede complementar el diagnóstico con un enema opaco.
Malrotación intestinal y vólvulo
La malrotación intestinal es un espectro de anomalías en la rotación y fijación del intestino, que puede tener consecuencias graves como el vólvulo intestinal.
La incidencia es aproximadamente del 1%, y la mayoría se diagnostican en el periodo neonatal. Se asocia a otras patologías como: gastrosquisis y onfalocele, hernia diafragmática congénita, y atresia duodenal o de vías biliares.
Esta malformación ocurre en el primer trimestre del embarazo, cuando no se produce la rotación del intestino al incorporarse al abdomen desde la cavidad celómica. Existen diferentes tipos de malrotaciones en función de donde se localiza el colon y el duodeno, siendo la más frecuente, aquella que tiene el ciego en el cuadrante superior derecho; en estos casos, suele asociarse a unas bandas peritoneales colecistoduodenocólicas, llamadas bandas de Ladd(11).
La clínica puede aparecer en cualquier momento de la vida, aunque la mayoría se diagnostica en el periodo neonatal por vómitos biliosos o por obstrucción duodenal (relacionado con las bandas de Ladd o complicaciones como el vólvulo intestinal). En los casos en los que el diagnóstico no es precoz, la clínica puede ser más difusa como: vómitos no biliosos intermitentes, dolor abdominal, diarrea, estreñimiento o fallo de medro. En ocasiones, debido a un vólvulo cronificado, se produce obstrucción linfática del meso y malabsorción intestinal. La exploración física suele ser normal, excepto en los casos en los que existe un vólvulo intestinal agudo, que presenta un cuadro clínico de: distensión abdominal, peritonismo y sangrado con las heces, debido a una necrosis intestinal(11,12).
El diagnóstico se puede realizar con un contraste digestivo que pone de manifiesto la malposición intestinal y puede mostrar una imagen característica en “sacacorchos” en el marco duodenal. También la ecografía, en ocasiones, se muestra útil para visualizar la inversión del eje mesentérico vascular.
El tratamiento es quirúrgico y es una emergencia en el caso de sospechar un vólvulo intestinal (Fig. 6).

Figura 6. Imagen quirúrgica de una malrotación intestinal con un vólvulo de intestino medio.
El procedimiento de Ladd es la técnica usada para corregir la malformación, que consiste en: revertir el vólvulo intestinal, resecar las áreas necrosadas, liberar las bandas de Ladd si están presentes y dejar el intestino en posición de “no rotación”, con todo el intestino delgado hacia la derecha y el grueso a la izquierda, para conseguir ampliar el meso del intestino y disminuir la posibilidad de vólvulo intestinal(13).
Atresia anorrectal
La malformación anorrectal (MAR) es una malformación congénita en la que la porción anorrectal se posiciona de forma anómala fuera del mecanismo esfinteriano del ano (completa o parcialmente).
Ocurre aproximadamente en 1 de cada 5.000 RNV y tiene un predominio masculino. Existe una clasificación de la MAR en función del sexo del paciente (Tabla I)(14).

Puede presentar malformaciones asociadas, como la asociación VACTERL; de estas, las más frecuentes son las anomalías genitourinarias.
El examen físico es la parte más importante a la hora del diagnóstico de estos pacientes. Así, en las “formas altas”, nos encontramos: un perine plano, meconiuria, un sacro corto y mala contracción esfinteriana. La presencia de un escroto bífido, en ocasiones, se asocia a fístulas prostáticas. En las niñas, la posición de la apertura de la fístula es importante tanto para el diagnóstico como para el pronóstico, pudiendo dividirse en: vestibular, perineal, vaginal o cloacal. Las “formas bajas” presentan una apertura de la fístula a nivel perineal, un periné bien conformado y, en ocasiones, lo que se conoce como asa de cubo, que es un repliegue cutáneo con forma de asa localizado a nivel perineal.
Para el diagnóstico, es muy útil la realización de una Rx abdomen-pelvis y la realización de un “invertograma” a las 24 horas del nacimiento (Rx lateral en posición decúbito prono con la pelvis ascendida), para ver la distancia entre el bolsón anorrectal y la piel. Otras pruebas complementarias preoperatorias obligatorias son: la ecocardiografía, la ecografía abdominal y la ecografía del canal medular, para descartar médula anclada y otras malformaciones genitourinarias asociadas.
El manejo inicial consiste en la colocación de: sonda nasogástrica, dieta absoluta, nutrición parenteral y antibioterapia profiláctica.
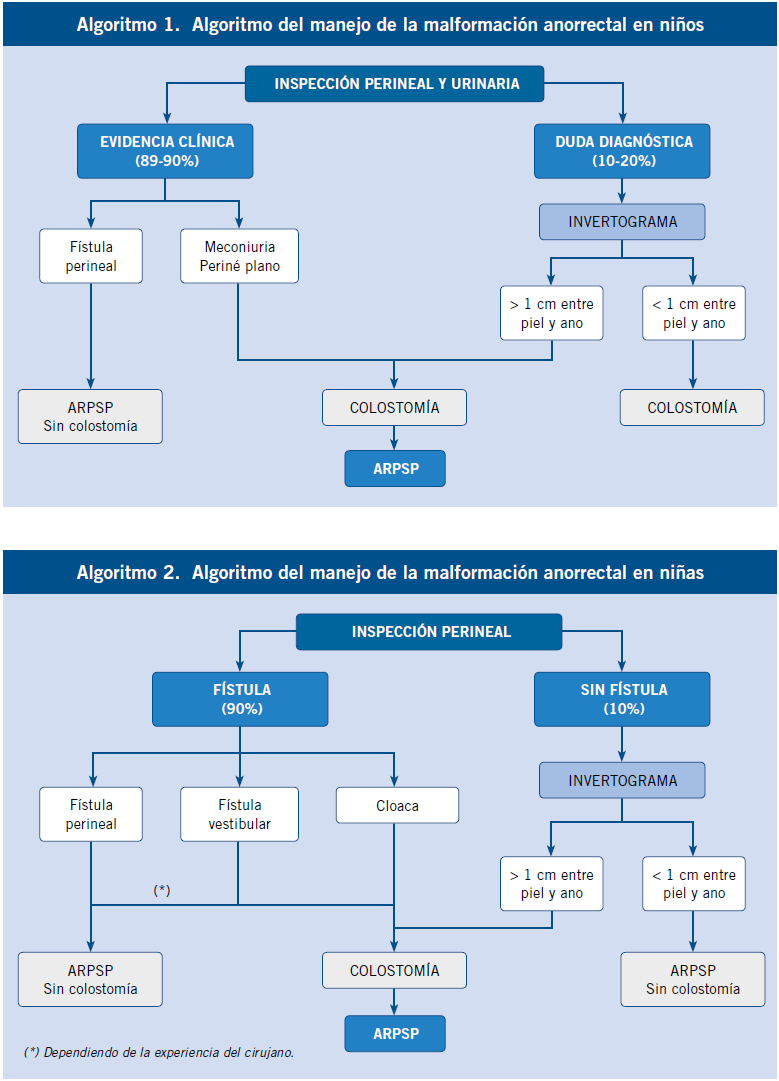
El manejo quirúrgico es variado y depende de la gravedad de la malformación. La reparación definitiva consiste en una anorrectoplastia sagital posterior (ARPSP), que consiste en la colocación del ano en su posición normal (Fig. 7).
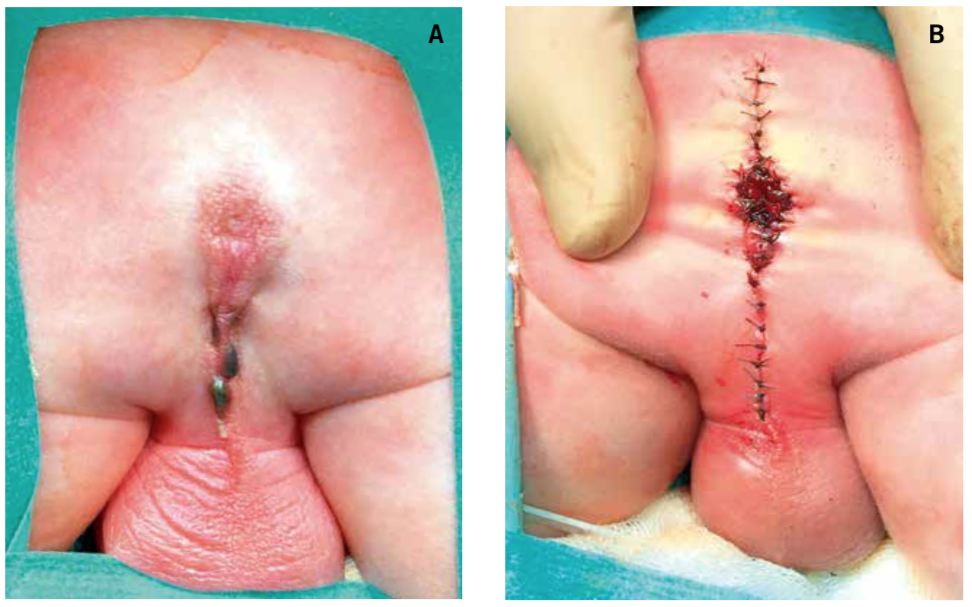 |
Figura 7.Imagen intraoperatoria de una malformación anorrectal. A. Atresia anal baja en la que se visualizan “perlas” de meconio en el rafe perianal. B.Imagen de la corrección de la malformación tras una anorrectoplastia sagital posterior.
Esta intervención, en ocasiones, se puede realizar en el periodo neonatal, pero en los casos más complejos, es necesaria la realización de un estoma de descarga y una ARPSP de forma diferida (Algoritmos 1 y 2)(14,15).
Con respecto al pronóstico de la MAR, la continencia es peor cuanto más alta es la malformación, así como mayor es la anomalía sacra. Cuanto más baja es la malformación, mejor es la continencia, aunque sufren mayor estreñimiento. Son pacientes que precisan de un seguimiento a largo plazo por parte de un cirujano pediátrico especializado en la patología colorrectal(16).
Duplicaciones digestivas
Se denominan duplicaciones digestivas aquellas formaciones quísticas que se desarrollan cercanas al tubo digestivo, generalmente sin comunicación con él. Su epitelio es digestivo y secretor y su localización es variable, pudiendo aparecer en cualquier tramo del tubo digestivo.
El diagnóstico, en general, es fortuito, por su presentación asintomática, aunque cada vez es más frecuente el diagnóstico prenatal. Ocasionalmente, se presentan con síntomas que son variados y dependen de la localización de la duplicación, pudiendo ser: dolor abdominal, disfagia, vólvulo intestinal, hemorragia digestiva, obstrucción intestinal, etc.
El diagnóstico se realiza mediante ecografía y resonancia magnética nuclear.
El tratamiento debe ser quirúrgico y consiste en la extirpación total o parcial de la masa, y, si es imposible, separar del intestino, puede necesitar una resección intestinal limitada (Fig. 8).
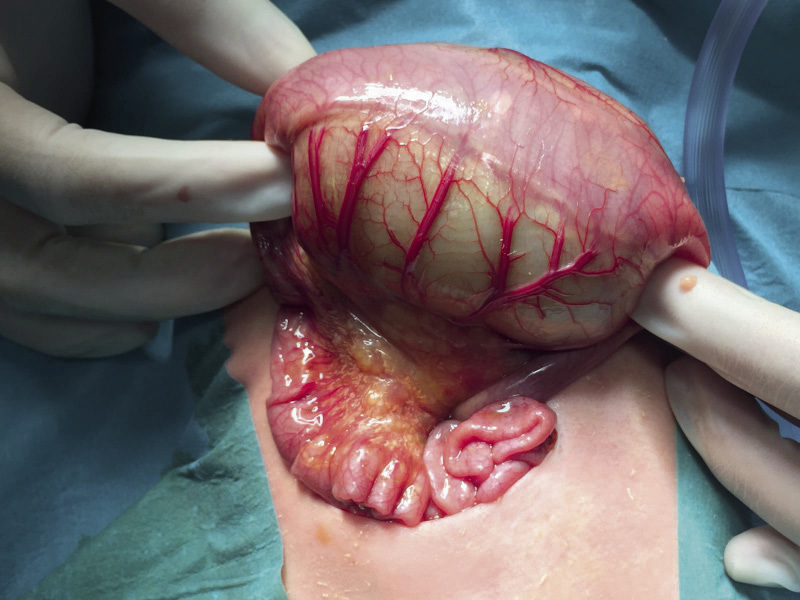
Figura 8. Imagen intraoperatoria de duplicación intestinal a nivel yeyunal.
Divertículo de Meckel
El divertículo de Meckel (DM) es una anomalía del tubo digestivo que consiste en una atresia incompleta del conducto onfalomesentérico que comunica el intestino con el saco vitelino.
Suele localizarse entre 50-75 cm de la válvula ileocecal, en el borde antimesentérico y puede tener tejido gástrico o pancreático, cuya secreción puede provocar úlcera y sangrado; de ahí, que sea una de las causas de hemorragia digestiva baja durante la infancia. Otras formas de debut son: la invaginación íleo-ileal difícilmente reductible, la diverticulitis de Meckel y más raramente como vólvulo intestinal (Fig. 9).
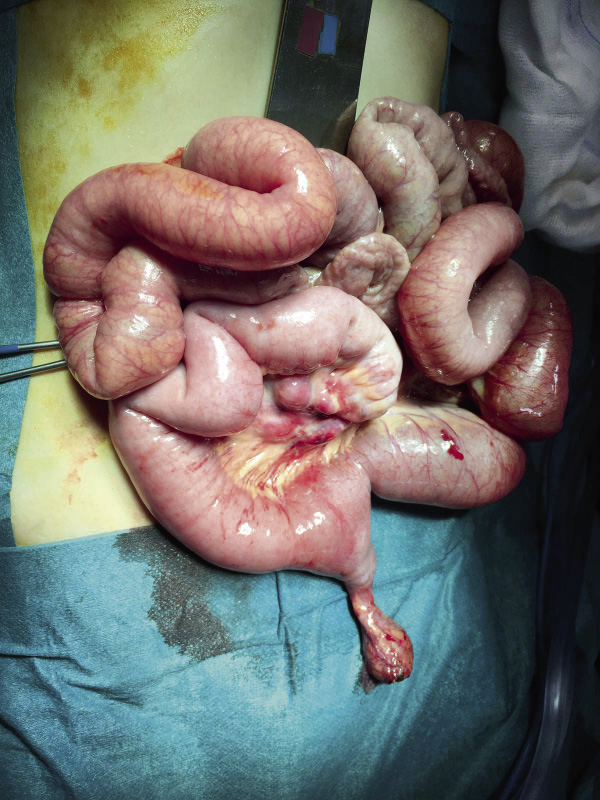
Figura 9. Imagen intraoperatoria de divertículo de Meckel.
La edad de diagnóstico típica son los dos años, con ligera predominancia masculina y la forma más frecuente de presentación es la hemorragia rectal no dolorosa aislada. Con respecto a la diverticulitis, se presenta con dolor abdominal e incluso peritonitis por perforación. También, puede presentar síntomas de obstrucción intestinal, si su forma de presentación es la invaginación o el vólvulo.
Su diagnóstico se realiza mediante una gammagrafía con Tc-99, que es una prueba segura y específica. La ecografía también puede ser de ayuda, sobre todo, en casos de diverticulitis e invaginación.
El tratamiento consiste en la extirpación quirúrgica del divertículo y la anastomosis término-terminal.
Defectos de la pared abdominal
Existen dos tipos de defectos mayores de la pared abdominal con diferente comportamiento y pronóstico, que son el onfalocele y la gastrosquisis.
El onfalocele es un defecto umbilical, producido por la regresión incompleta de las vísceras abdominales a la cavidad. Como consecuencia, persiste un defecto cubierto por un saco con tres capas: el peritoneo, la gelatina de Wharton y la membrana amniótica. Su tamaño es variable, denominándose “minor” si es menor de 5 cm de diámetro y “maior” si es de más de 5 cm. Ocurre en 1/5.000 RNV y suele asociarse a otras anomalías, a cromosomopatías y al síndrome de Beckwith-Wiedeman (Fig. 10)(17,18).

Figura 10. Imagen de paciente con onfalocele.
La gastrosquisis consiste en un fracaso de la vascularización de la pared abdominal, lo cual se manifiesta con un defecto paraumbilical derecho que suele tener un tamaño de 2-4 cm, con una inserción correctal del ombligo. Por dicho orificio protruyen vísceras no cubiertas. Ocurre en 1/2.000 RNV y se asocia a: edad baja materna, uso de drogas y, en ocasiones, aunque no tiene asociaciones a otras anomalías, sí que se puede acompañar de atresias intestinales, debido a procesos isquémicos (Fig. 11)(18).

Figura 11. Imagen intraoperatoria de gastrosquisis.
El diagnóstico prenatal es muy frecuente y, tras el nacimiento, las pruebas a realizar son: ecografía y ecocardiograma. Al nacimiento, hay que descomprimir el estómago con SNG y cubrir el defecto para la protección de las vísceras y para disminuir la pérdida de calor corporal. Hay que añadir antibioterapia. El tratamiento quirúrgico consiste en la reintroducción de las vísceras y cierre del defecto abdominal. En ocasiones, esto no es posible porque hay una falta de espacio en el abdomen; por lo que, ocasionalmente, se necesita la colocación de un silo para que la introducción de las vísceras sea progresiva, disminuyendo así la posibilidad de síndrome compartimental (Fig. 12).

Figura 12. Imagen intraoperatoria de una gastrosquisis con un silo preformado.
Con respecto a su pronóstico, habitualmente, tienen retraso en la introducción de nutrición enteral, debido a la dismotilidad intestinal. Aunque la supervivencia es buena, existen casos de intestino corto secundario a estos procesos, sobre todo, la gastrosquisis, por episodios isquémicos tanto intraúteros como postnatales(19).
Bibliografía
1. Spaggiari E, Faure G, Rousseau V, Sonigo P, Millischer-Bellaiche AE, Kermorvant-Duchemin E, et al. Performance of prenatal diagnosis in esophageal atresia. Prenat Diagn. 2015; 35: 888-93. doi:10.1002/pd.4630.
2. Rubio EI, Blask AR, Badillo AT, Bulas DI. Prenatal magnetic resonance and ultrasonographic findings in small-bowel obstruction: imaging clues and postnatal outcomes. Pediatr Radiol. 2017; 47: 411-21. doi:10.1007/s00247-016-3770-0.
3. Catania VD, Briganti V, Di Giacomo V, Miele V, Signore F, de Waure C, et al. Fetal intra-abdominal cysts: accuracy and predictive value of prenatal ultrasound. J Matern Fetal Neonatal Med. 2016; 29: 1691-9. doi:10.3109/14767058.2015.1059812.
4. Shieh HF, Jennings RW. Long-gap esophageal atresia. Semin Pediatr Surg. 2017; 26: 72-7, doi:10.1053/j.sempedsurg.2017.02.009.
5. Peters RT, Ragab H, Columb MO, Bruce J, MacKinnon RJ, Craigie RJ. Mortality and morbidity in oesophageal atresia. Pediatr Surg Int. 2017; 33: 989-94, doi:10.1007/s00383-017-4124-1.
6. Sfeir R, Michaud L, Sharma D, Richard F, Gottrand F. National Esophageal Atresia Register. Eur J Pediatr Surg. 2015; 25: 497-9. doi: 10.1055/s-0035-1569466.
7. Gottrand M, Michaud L, Sfeir R, Gottrand F. Motility, digestive and nutritional problems in Esophageal Atresia. Paediatr Respir Rev. 2016; 19: 28-33. doi.org/10.1016/j.prrv.2015.11.005.
8. Zani A, Yeh JB, King SK, Chiu PP, Wales PW. Duodeno-duodenostomy or duodeno-jejunostomy for duodenal atresia: is one repair better than the other? Pediatr Surg Int. 2017; 33: 245-8. doi:10.1007/s00383-016-4016-9.
9.** Millar AJW, Rode H, Cywes S. Intestinal atresia and stenosis. In: Ashcraft KW, Holder TM, eds. Pediatric Surgery, Saunders, 2000. P 406-24.
10. Dalla Vecchia LK, Grosfeld JL, West KW, Rescorla FJ, Scherer LR, Engum SA. Intestinal atresia and stenosis: a 25-year experience with 277 cases. Arch Surg. 1998; 133: 490-6.
11. Strouse PJ. Malrotation. Semin Roentgenol. 2008; 43: 7-14, doi:10.1053/j.ro.2007.08.002.
12.** Millar AJ, Rode H, Cywes S. Malrotation and volvulus in infancy and childhood. Semin Pediatr Surg. 2003; 12: 229-36.
13. Bass KD, Rothenberg SS, Chang JH. Laparoscopic Ladd’s procedure in infants with malrotation. J Pediatr Surg. 1998; 33: 279-81.
14. Levitt MA, Peña A. Anorectal malformations. OJRD. 2007; 2: 33. doi:10.1186/1750-1172-2-33.
15.** Rintala RJ, Pakarinen MP. Imperforate anus: long- and short-term outcome. Semin Pediatr Surg. 2008; 17: 79-89. doi:10.1053/j.sempedsurg.2008.02.003.
16. Levitt MA, Peña A. Outcomes from the correction of anorectal malformations. Curr Opin Pediatr. 2005; 17: 394-401.
17. Watanabe S, Suzuki T, Hara F, Yasui T, Uga N, Naoe A. Omphalocele and gastroschisis in newborns: over 16 years of experience from a single clinic. J Neonat Surg. 2017; 6: 27-30, doi:10.21699/jns.v6i2.530.
18. Kelly KB, Ponsky TA. Pediatric abdominal wall defects. Surg Clin North Am. 2013; 93: 1255-67. doi:10.1016/j.suc.2013.06.016.
19. Gamba P, Midrio P. Abdominal wall defects: prenatal diagnosis, newborn management, and long-term outcomes. Semin Pediatr Surg. 2014; 23: 283-90. doi:10.1053/j.sempedsurg.2014.09.009.
Bibliografía recomendada
– Coran A, Brusch SW, Kunisaki SM. Esophageal Atresia and Tracheoesophageal Fistula. Pediatric Digestive Surgery; 2017. p. 169-82.
Buena revisión del tema de la atresia de esófago, en la que se abordan todos los conceptos relacionados con la patología, siendo de lectura sencilla y con conceptos básicos.
– Varlet F. Vermersch S, Bustangi N, López M, Scalabre A. Meckel’s Diverticulum. Pediatric Digestive Surgery; 2017. p. 269-77.
Revisión básica, pero completa, sobre todo, el espectro del divertículo de Meckel en un libro de referencia de cirugía abdominal pediátrica, que debería de ser de cabecera para los especialistas de Pediatría.
– Little DC, Smith SD. Malrotation. Ashcraft’s Pediatric Surgery; 2010. p. 416-24.
Libro de referencia en cirugía pediátrica, que desarrolla de forma exhaustiva toda la patología quirúrgica en niños y que, en este capítulo, muestra la visión más completa sobre la malrotación intestinal.
– Partridge E, Hedrick H. Duodenal Atresia and Stenosis. Rickham’s Neonatal Surgery; 2018. p. 675-81. doi: 10.1007/978-1-4471-4721-3.
Capítulo sobre la atresia duodenal, con una revisión completa desde el punto de vista del diagnóstico y del tratamiento.
– Levitt M, Wood R. Anorectal Malformations. Rickham’s Neonatal Surgery; 2018. p. 629-38. doi: 10.1007/978-1-4471-4721-3.
Capítulo de malformaciones anorrectales en un libro de cirugía neonatal escrito por expertos a nivel mundial, explicando dicho capítulo de forma comprensiva.
| Caso clínico |
|
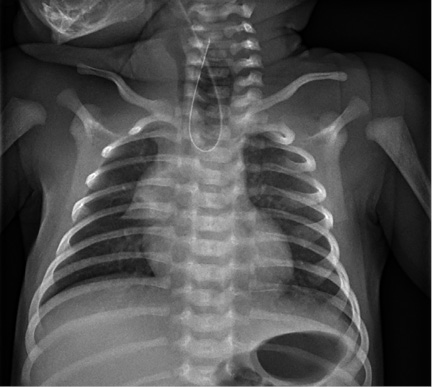
Recién nacido a término (41 semanas + 3 días) de peso adecuado para la edad gestacional. Peso. 3.020 g; Longitud: 49 cm y PC: 35,5 cm. Al nacimiento, se administra profilaxis ocular y antihemorrágica. Realiza primera diuresis y expulsión de meconio en las primeras 24 horas. Ingresa a las 12 horas de vida, procedente de maternidad por: regurgitaciones frecuentes de contenido mucoso-alimentario, sialorrea, náuseas y distrés respiratorio (Silverman de 2). Al ingreso, se objetiva saturación de 86% y se intenta, mediante colocación de sonda, comprobar la permeabilidad de la comunicación orogástrica, sin éxito. Se realiza radiografía de tórax en la que presenta bucle de la misma, a nivel del tercio superior esofágico. Exploración física Buen estado general. Buena coloración de piel y mucosas. No lesiones cutáneas. Mínimo tiraje subcostal e intercostal. ACP adecuada, entrada de aire bilateral con algún crepitante bilateral. Rítmico, sin soplos. Abdomen blando, no excavado. Abundantes secreciones en región oral. Neurológico: vital y reactivo. Clavículas íntegras, no anomalías de extremidades. Genitales masculinos normales, con ano perforado. Pruebas complementarias • Radiografía de tórax: ausencia de paso de la sonda nasogástrica, haciendo un bucle en el tercio proximal del esófago. Doce pares de costillas. Resto de la radiografía normal (Fig. 13). • Ecografía abdominal: ligero bloqueo tubular renal. Ligera-moderada cantidad de líquido subhepático derecho y periesplénico (mayor en el lado derecho). • Ecocardiograma: corazón anatómica y estructuralmente normal. Ductus Arterioso persistente pequeño sin repercusión. Foramen oval permeable. • Ecografía cerebral: mínimas imágenes de germinolisis. Pequeño quiste de plexo coroideo derecho. • Interconsulta a genética: hallazgos sugestivos de atresia de esófago aislada, no sindrómica. Evolución Se interviene a las 24 horas de vida sin incidencias, realizándose un cierre de fístula traqueoesofágica y una anastomosis término-terminal esofágica. Se deja drenaje torácico con escaso débito, retirado a los 2 días de la intervención. Se deja una sonda nasogástrica transanastomótica, por la que se inicia nutrición enteral trófica el 6º día de vida. Tras comprobar la estanqueidad de la anastomosis, se procede a introducir alimentación oral.
Figura 13.
|