 |
| Temas de FC |
M. Serrano Valls, C. de Lucas Collantes, C. Aparicio López
Servicio de Nefrología Pediátrica, Hospital Infantil Universitario Niño Jesús, Madrid
| Resumen
Los quistes renales son un hallazgo radiológico frecuente, tanto en niños como en adultos. Pueden ser adquiridos o formar parte de una enfermedad hereditaria, aparecer de forma aislada, múltiple o en el contexto de síndromes complejos o malformaciones congénitas renales. En los últimos años, se ha establecido una relación entre la alteración funcional de los cilios primarios y la formación de quistes renales, lo que nos ha permitido conocer mejor la patogenia de estas enfermedades. En este artículo, revisaremos las patologías renales quísticas más relevantes para favorecer su comprensión por parte del pediatra de Atención Primaria y facilitar tanto su detección precoz como su seguimiento posterior. |
| Abstract
Renal cysts are a common radiological finding in both adults and children. Cyst can be simple or multiple. They may be acquired, part of complex hereditary syndromes or renal congenital malformations. In the last years an association of the dysfunction of primary cilia and the formation of renal cyst has been described, which enables us to better understand the pathogenesis of this disease. In this article we review the most relevant renal cystic diseases so the primary care pediatrician can be more familiar with them and thereby do an earlier diagnosis as well as a better follow-up. |
Palabras clave: Quistes renales; Riñón poliquístico; Displasia renal multiquística; Ciliopatías
Key words: Renal cysts; Polycystic kidney; Multicystic dysplastic kidney; Ciliopathies
Pediatr Integral 2017; XXI (8): 541 –548
Enfermedades quísticas renales
Introducción
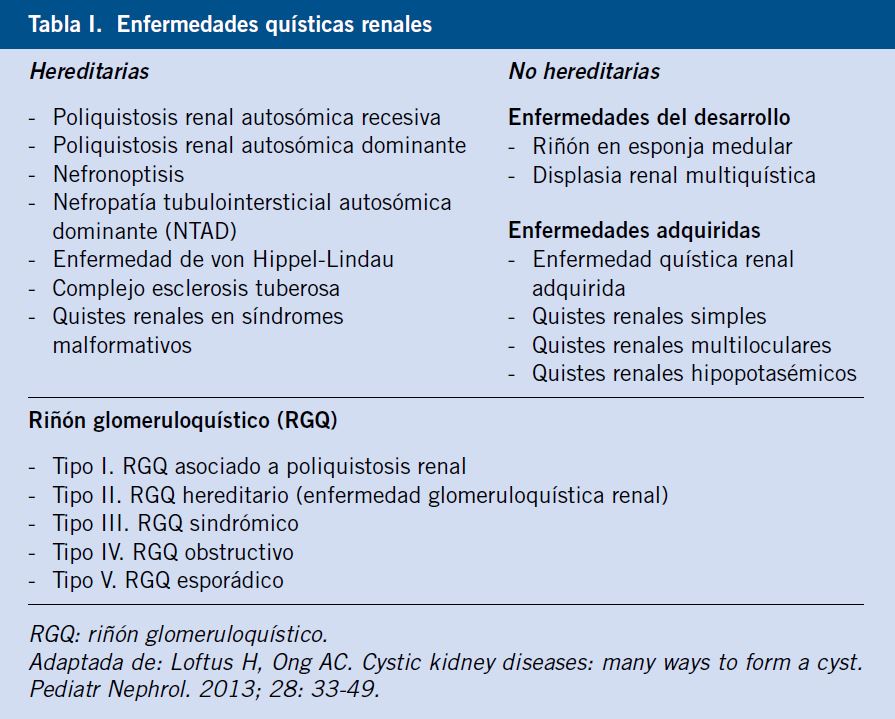
Se denomina quiste renal a la formación consistente en una dilatación tubular de tamaño 4 veces superior al normal(1). Las enfermedades quísticas renales comprenden un grupo de patologías muy heterogéneo, que engloba tanto enfermedades de origen genético como adquirido, con una edad de presentación muy variable, que abarca desde la etapa prenatal hasta la edad adulta. Suponen la causa hereditaria más frecuente de enfermedad renal terminal (ERT) en la población pediátrica. Existen numerosas clasificaciones de estas enfermedades, según diversos criterios, como su origen o el número de quistes. En la tabla I, se incluye la clasificación propuesta por Loftus en el año 2013(2). Revisaremos en este artículo, las patologías quísticas renales más relevantes atendiendo a su origen genético o adquirido.
Patogenia
La alteración de la estructura y la función de los cilios primarios parece un factor común en la patogenia de las enfermedades quísticas renales.
Los cilios primarios son organelas microtubulares localizadas en la superficie de la mayoría de las células del organismo, que cumplen funciones de recepción de estímulos y de señalización celular. Las proteínas asociadas a fenotipos renales quísticos se encuentran localizadas, en su mayoría, en los cilios primarios o los centrosomas asociados a estos. Aunque la patogenia no está clara, se piensa que la función de los cilios primarios es fundamental para el mantenimiento del diámetro tubular durante su elongación en el desarrollo fetal y, posteriormente, en la reparación del daño tubular siendo, la alteración de dicha función, clave en el desarrollo de quistes renales. La presencia de cilios en otros tejidos del organismo también podría explicar alguna de las manifestaciones extrarrenales asociadas a las enfermedades quísticas renales. Por este motivo, la mayoría de dichas enfermedades se engloban en el grupo denominado ciliopatías(1,2).
Enfermedades quísticas renales hereditarias (Tabla II)
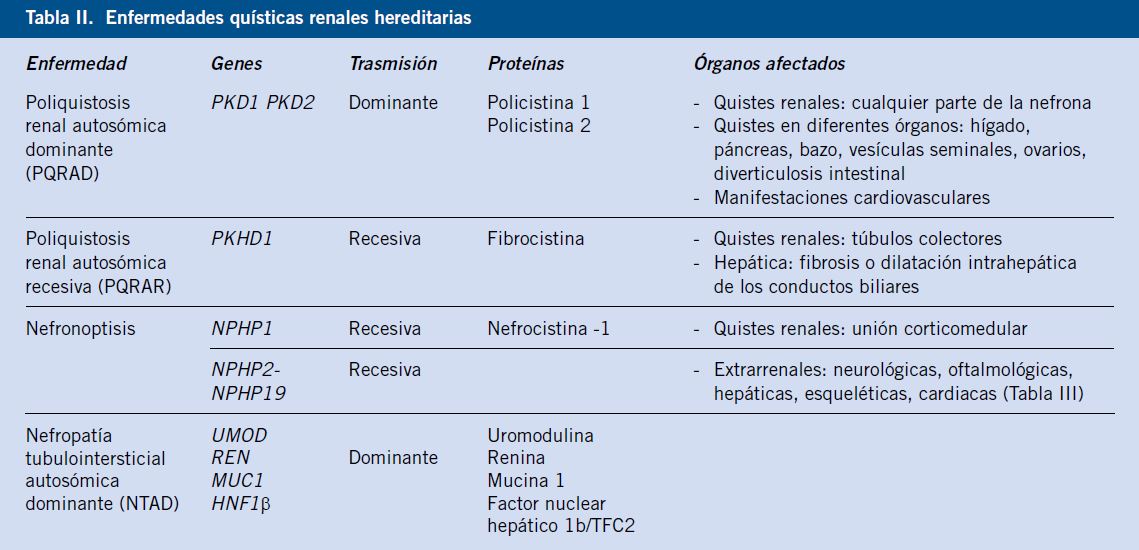
Poliquistosis renal autosómica recesiva (PQRAR)
Se caracteriza por insuficiencia renal y afectación hepática con hipertensión portal secundaria, con debut en la primera infancia y evolución hacia el trasplante renal o hepatorrenal.
La poliquistosis renal autosómica recesiva (PQRAR) es una enfermedad quística renal causada por mutaciones en el gen PKHD1 (cromosoma 6p21.1-p12), que codifica la proteína fibrocistina. Esta proteína, cuya función exacta es desconocida, se localiza en los cilios primarios y en los cuerpos basales de las células de los túbulos colectores renales, así como en otros órganos, como hígado y páncreas.
El defecto de la fibrocistina da lugar principalmente al desarrollo de quistes renales, localizados en los túbulos colectores, y de fibrosis hepática o dilatación intrahepática de los conductos biliares (enfermedad de Caroli).
Tiene una incidencia de 1:20.000 recién nacidos vivos, sin diferencias entre sexos(2).
La PQRAR presenta una gran variabilidad clínica. En la mayoría de los casos, se observan manifestaciones intraútero o al nacimiento, cursando las formas neonatales más severas con: oligoamnios, hipoplasia pulmonar secundaria y facies típica, lo que se conoce como secuencia de Potter.
Por lo general, los pacientes afectos de PQRAR presentan un defecto de concentración urinaria, que se manifiesta ya desde la etapa neonatal en forma de poliuria y polidipsia secundaria a esta. La hipertensión arterial (HTA) es otro hallazgo precoz y puede ser severa y de difícil control, asociando en los casos más graves, hipertrofia del ventrículo izquierdo e incluso insuficiencia cardiaca congestiva. Pueden asociar, además, infecciones urinarias de repetición.
La afectación hepática se caracteriza por fibrosis del espacio periportal y enfermedad de Caroli (dilatación intrahepática de los conductos biliares), que conlleva con frecuencia la aparición de hipertensión portal (HTP). Clínicamente, se presentan con: hepatoesplenomegalia, hiperesplenismo con pancitopenia y varices esofágicas con riesgo de sangrado. El daño hepatocelular es infrecuente y la función hepática no suele verse afectada. Sin embargo, sí que se puede ver deteriorada de forma secundaria a colangitis bacterianas de repetición, que suponen una de las complicaciones hepáticas más frecuentes y graves(1-4).
Los pacientes que debutan en la adolescencia, o en edades más tardías, presentan una afectación clínica de predominio hepático que, en ocasiones, puede ser la única manifestación.
La PQRAR conlleva una elevada morbimortalidad con evolución hacia enfermedad renal terminal (ERT), que se alcanza entre los 5 y los 15 años de edad(1). Se estima que entre un 23-30% de los neonatos afectos fallece en el primer año de vida, como consecuencia de la insuficiencia respiratoria secundaria a hipoplasia pulmonar(2,3). Para aquellos que sobreviven al primer año, la supervivencia se estima en un 82%(2,3). En estos pacientes, se ha descrito una mejoría transitoria del filtrado glomerular en los primeros 2 años de vida, antes de iniciar un descenso progresivo hasta alcanzar la ERT(4).
Diagnóstico
Se basa en los hallazgos clínicos y en las pruebas de imagen. En la ecografía al nacimiento, se observan unos riñones aumentados de tamaño e hiperecogénicos, con pérdida de la diferenciación corticomedular y, en ocasiones, con pequeños quistes (< 15-20 mm). La ecografía prenatal puede orientar ya el diagnóstico en algunos casos, mostrando aumento del tamaño y ecogenicidad renal y oligoamnios. Los macroquistes característicos de la poliquistosis renal autosómica dominante (PQRAD) pueden aparecer con la evolución de la enfermedad en niños mayores y adolescentes(1,4). La ausencia de quistes en el estudio ecográfico de los padres, puede ser de utilidad en el diagnóstico diferencial con la PQRAD.
El hígado puede ser ecográficamente normal en niños pequeños o mostrar hepatomegalia con incremento de la ecogenicidad del parénquima, signos de HTP, esplenomegalia y, ocasionalmente, quistes hepáticos y de colédoco(1-4).
El estudio anatomo-patológico solo se realiza en casos excepcionales. El diagnóstico genético es laborioso, ya que el gen PKHD1 es largo y complejo, con posibilidad de un gran número de mutaciones y polimorfismos(3,4).
Tratamiento
No existe actualmente ningún tratamiento específico para la PQRAR, siendo su tratamiento sintomático y de soporte. El periodo neonatal es el más crítico y requiere, por lo general, el ingreso en unidades de cuidados intensivos por insuficiencia respiratoria secundaria a hipoplasia pulmonar, precisando, en ocasiones, soporte con ventilación mecánica. La nefrectomía uni o bilateral para intentar mejorar la dinámica respiratoria no se recomienda de forma rutinaria, aunque puede considerarse en casos concretos(5).
El tratamiento conservador de la enfermedad renal incluye el manejo de la HTA, siendo de elección los inhibidores de la enzima convertidora de angiotensina (IECAs) o los antagonistas de los receptores de angiotensina (ARA II)(5), y el soporte nutricional desde la etapa neonatal que, en ocasiones, requiere el empleo de gastrostomía o sonda nasogástrica. A medio plazo, una vez superados los dos primeros años de vida, puede ser preciso el tratamiento precoz con hormona de crecimiento(1). Asimismo, se deben tratar de forma precoz, las complicaciones asociadas (infecciones urinarias, colangitis bacteriana, sangrado de varices esofágicas). La utilidad de la profilaxis antibiótica para prevenir las colangitis de repetición es controvertida y no se recomienda salvo en periodos cortos de 6-12 semanas, tras un episodio de colangitis o inmediatamente después del trasplante(5).
A largo plazo, la mayoría de los pacientes serán subsidiarios de trasplante renal aislado o trasplante hepatorrenal. El pronóstico del trasplante es bueno, ya que la enfermedad no recidiva en el injerto.
En el momento actual, se están estudiando nuevas dianas terapéuticas específicas (antagonistas del receptor de vasopresina 2, inhibidores de tirosín-quinasa específicos para factor de crecimiento epidérmico…), aunque no existe evidencia suficiente que las respalde como un tratamiento consolidado.
Poliquistosis renal autosómica dominante (PQRAD)
La aparición de macroquistes renales, que sustituyen al parénquima renal sano, deriva en insuficiencia renal terminal. Aunque suele debutar en la tercera o cuarta década de la vida, también existen formas infantiles.
La poliquistosis renal autosómica dominante (PQRAD) es un trastorno multisistémico caracterizado por la aparición de quistes en diferentes órganos. Se considera la enfermedad renal hereditaria más frecuente, con una incidencia de 1 de cada 400-1.000 recién nacidos vivos(2,3,6).
Es el resultado de mutaciones en dos genes diferentes, PKD1 (cromosoma 16p13.3) y PKD2 (cromosoma 4q21-q23), que codifican dos proteínas localizadas en los cilios primarios y en otros compartimentos celulares, como el retículo endoplásmico, denominadas policistina 1 y policistina 2 (o TRPP2), respectivamente(6,7). La alteración de cualquiera de estas dos proteínas da lugar a fenotipos similares caracterizados por la formación de quistes renales que, al aumentar en tamaño y número, sustituyen progresivamente el parénquima renal sano, desarrollando en su evolución, inflamación y fibrosis intersticial. Estos quistes, a diferencia de lo que ocurre en la PQRAR, pueden afectar a cualquier parte de la nefrona y no solo a los túbulos colectores. Con frecuencia, también se desarrollan quistes en otros órganos.
Las mutaciones en el gen PKD1 suponen un 80-85% de los casos de PQRAD y dan lugar a fenotipos más graves que aquellos ocasionados por mutaciones en PKD2, que suponen el 15-20% restante(1,3,6).
La mayoría de los pacientes debutan clínicamente en la tercera o cuarta década de la vida; sin embargo, puede detectarse también en la población pediátrica, bien por el hallazgo de quistes renales en niños asintomáticos con antecedentes familiares, o bien por la existencia de formas graves de debut precoz en la infancia denominadas VEO (Very Early Onset), que suponen hasta un 2-5% de los casos(3). Se han descrito incluso formas de presentación neonatal muy similares a la PQRAR, con una elevada morbimortalidad e insuficiencia renal en los primeros años de vida(6). Estas formas de debut precoz tienen, además, un elevado riesgo de recurrencia dentro de la misma familia(3,6).
Las manifestaciones renales, tanto en población pediátrica como en adultos, pueden incluir: HTA, hematuria, hipostenuria, litiasis renal, infecciones urinarias, dolor abdominal en relación con infección o hemorragia de los quistes, y finalmente, progresión a ERT.
La HTA es uno de los hallazgos más frecuentes, apareciendo de manera precoz, incluso antes de que se produzca una reducción significativa del filtrado glomerular, y supone el principal factor de progresión hacia la insuficiencia renal, además de estar asociada a un mayor riesgo de complicaciones cardiovasculares(3,6). La hipertrofia de ventrículo izquierdo aparece en un 44% de estos pacientes y se asocia a mortalidad de causa cardiovascular(3). La progresión a ERT se suele producir en la edad adulta avanzada, siendo más precoz en las formas secundarias a mutaciones en el gen PKD1, que suelen alcanzar la ERT una media de 20 años antes que aquellas producidas por mutaciones en el gen PKD2 (54 vs 74 años de media)(1,3,6).
Las manifestaciones extrarrenales son menos frecuentes en la población pediátrica y engloban la aparición de quistes en diferentes órganos (hígado, páncreas, bazo, vesículas seminales, ovarios, diverticulosis intestinal) y manifestaciones cardiovasculares como: la aparición de aneurismas cerebrales, dilatación de la raíz aórtica o prolapso de la válvula mitral. Los quistes hepáticos son la manifestación extrarrenal más frecuente. Suelen ser benignos y, por lo general, no afectan a la función hepática. Aunque la PQRAD afecta por igual a ambos sexos, la afectación hepática es más frecuente, severa y precoz en las mujeres, mientras que la afectación renal suele ser más grave en varones(3,6). Los aneurismas cerebrales aparecen en aproximadamente el 8% de los individuos afectos y tienden a la agrupación familiar. La ruptura de dichos aneurismas, aunque rara, también se ha descrito en la población pediátrica(3).
Existe, además, una forma grave de enfermedad poliquística asociada a esclerosis tuberosa, con debut en la primera infancia. Se denomina síndrome de genes contiguos TSC2/PKD1, ya que ocurre como consecuencia de deleciones que afectan tanto a PKD1 como al gen TSC2 (tuberina), que se encuentran adyacentes en el genoma. Debe sospecharse en pacientes diagnosticados de PQRAD con debut precoz, sin antecedentes familiares de la enfermedad, especialmente aquellos con una clínica compatible con esclerosis tuberosa. En estos casos, se recomienda la realización de una RM craneal y estudio cardiológico que nos permita descartar esclerosis tuberosa, pudiéndose confirmar posteriormente el diagnóstico mediante estudio genético(1,6,8).
Diagnóstico
Se basa principalmente en los hallazgos ecográficos. Los riñones pueden aparecer aumentados de tamaño, con presencia de quistes, distribuidos entre corteza y médula, y escasa diferenciación corticomedular. La presencia de enfermedad en el estudio ecográfico de los padres apoya el diagnóstico, especialmente en aquellos casos de debut precoz en los que se plantea el diagnóstico diferencial con PQRAR y otros síndromes malformativos. Sin embargo, la ausencia de alteraciones en dicho estudio no excluye el diagnóstico de PQRAD, ya que el 8-10% de los casos son mutaciones de novo(1,3,7).
El diagnóstico genético no se realiza de forma sistemática, debido al elevado polimorfismo de PKD1, que dificulta su realización e interpretación, y a su elevado coste económico(2,7). Se reserva para:
• La identificación de donante renal vivo emparentado, menor de 40 años, en familias afectas (la ausencia de alteraciones ecográficas en mayores de 40 años podría excluir la presencia de enfermedad[1-3]).
• Solapamiento fenotípico con otras enfermedades e historia familiar negativa para PQRAD.
• Consejo genético en familias con historia de PQRAD de debut precoz (riesgo del 45% de recidiva en próximos embarazos[2,6,7]).
• En familias con antecedentes de ruptura de aneurismas cerebrales.
El cribado mediante estudio genético o ecográfico en niños asintomáticos con riesgo de desarrollar PQRAD no se recomienda, dada la ausencia de tratamientos específicos. Sin embargo, algunos autores recomiendan la monitorización de las cifras de tensión arterial en estos pacientes y la instauración precoz de tratamiento antihipertensivo, ya que la HTA supone el principal factor de progresión a insuficiencia renal(2,7). Se recomienda asimismo el cribado de aneurismas cerebrales mediante resonancia magnética (RM) en pacientes de riesgo a partir de los 20 años de edad, especialmente si existe historia familiar de ruptura de los mismos(3). En la tabla III, se describen las principales diferencias entre PQRAD y PQRAR.

Tratamiento
El Tolvaptán, antagonista del receptor V2 de la vasopresina, es el único fármaco aprobado por la Agencia Europea del Medicamento para el tratamiento de la PQRAD en adultos, con ERC (enfermedad renal crónica) en estadios 1-3 y progresión rápida de la enfermedad. Este fármaco ha demostrado una disminución del volumen renal total, con enlentecimiento de la tasa de declive del filtrado glomerular estimado en estos pacientes(9-10). Por el momento, el Tolvaptán no está aprobado para su uso en la población pediátrica. Actualmente, existe un ensayo clínico multicéntrico fase III en curso que pretende evaluar su seguridad y eficacia en niños y adolescentes con diagnóstico de PQRAD(11).
Por tanto, el tratamiento en dicha población continúa siendo un tratamiento de soporte, orientado al control precoz de la HTA, así como de las posibles complicaciones (ITU, litiasis, infección o ruptura de los quistes), y al tratamiento de la ERC.
La nefrectomía unilateral de uno de los riñones nativos debe considerarse previa al trasplante en el caso de que el tamaño de dicho riñón impida la colocación adecuada del injerto, o en aquellos pacientes con hemorragias o infecciones intraquísticas de repetición(1,6).
Nefronoptisis
Nefropatía tubulointersticial que debuta con frecuencia en forma de insuficiencia renal avanzada en la adolescencia.
La nefronoptisis es una nefropatía tubulointersticial de herencia autosómica recesiva, que supone la causa hereditaria más frecuente de ERT en niños y adolescentes(1,12,13), con una incidencia de 1:50.000-1.000.000 de recién nacidos vivos(2,12,13).
En la actualidad, se han descrito hasta 19 genes relacionados con esta patología (NPH1-19), siendo la mutación en NPHP1 la más frecuente (20-25%). Sin embargo, en el 60-70% de los casos no se puede identificar el gen causante de la enfermedad(13). Las proteínas codificadas por dichos genes se localizan principalmente en los cilios primarios y el centrosoma(12,13).
Se diferencian 3 formas clínicas de nefronoptisis, en función de la edad de presentación. La forma clásica o juvenil es la más frecuente. Los primeros síntomas se suelen desarrollar a partir de los 4-6 años de edad, mientras que la ERT aparece a una edad media de 13 años. En la forma del adolescente, la ERT se alcanza a una edad media de 19 años; y en la forma infantil, que es la más severa, la progresión a ERT ocurre antes de los 4 años de edad, con manifestaciones incluso intraútero.
Todos los subtipos cursan con una clínica similar, donde destaca la disminución de la capacidad de concentración urinaria con pérdida de sodio en orina, que se traduce en poliuria y polidipsia, enuresis secundaria y riesgo de deshidratación. Suelen presentar, además: retraso del crecimiento, anemia normocítica normocrómica y las manifestaciones típicas de la enfermedad renal crónica (ERC). La tensión arterial suele mantenerse en rangos normales, debido a la pérdida urinaria de sodio, hasta que se alcanza un estado de insuficiencia renal, a excepción de las formas infantiles, que suelen cursar con hipertensión arterial de difícil control(1,2,12).
Hasta en un 10-20% de los casos, se asocian manifestaciones extrarrenales (neurológicas, oftalmológicas, hepáticas, esqueléticas, cardiacas), dando lugar a diferentes síndromes clínicos (Tabla IV).

Diagnóstico
Suele ser tardío, ya que los síntomas son poco llamativos hasta estadios muy avanzados de insuficiencia renal. En el estudio ecográfico, se observan: unos riñones de tamaño normal o reducido, hiperecogénicos, con pobre diferenciación córtico-medular y microquistes en la unión córtico-medular.
La biopsia renal será necesaria en aquellos casos en los que no se pueda identificar una causa genética(1,2,12,14). El aspecto histológico se distingue por un característico engrosamiento irregular de la membrana basal glomerular y fibrosis intersticial con atrofia tubular. Puede observarse también la presencia esporádica de quistes en la unión córtico-medular.
La forma infantil presenta un aspecto diferente, con riñones aumentados de tamaño y microquistes corticales, y ausencia típica de las alteraciones en la membrana basal tubular.
Tratamiento
Consiste en el manejo conservador de la ERC, con énfasis especial en el aporte de líquidos. El trasplante renal suele tener un buen pronóstico, ya que la nefronoptisis no recidiva en el injerto.
Nefropatía tubulointersticial autosómica dominante (NTAD)
Entidad de herencia autosómica dominante, poco frecuente, que suele debutar en la edad adulta. Debe sospecharse en pacientes jóvenes con hiperuricemia y/o gota, que progresan a insuficiencia renal.
El término nefropatía tubulointersticial autosómica dominante (NTAD) ha sido establecido recientemente en las últimas guías KDIGO, sustituyendo a lo que previamente se conocía como enfermedad quística medular(15). Engloba un grupo heterogéneo de nefropatías tubulointersticiales hereditarias poco frecuentes, que tienen en común la fibrosis tubulointersticial y la progresión lenta a ERT. Suelen debutar en la edad adulta, aunque pueden presentarse en la población pediátrica. La presencia de quistes es inconstante y no constituye un criterio necesario para el diagnóstico.
La NTAD se clasifica en 5 subtipos en función de la alteración genética subyacente(1,15):
1. Mutaciones en gen UMOD (uromodulina o proteína de Tamm-Horsfall): son las más frecuentes. Producen formas de nefropatía tubulointersticial asociadas a hiperuricemia hipouricosúrica y gota en el adolescente, con evolución a insuficiencia renal terminal entre los 25 y 70 años.
2. Mutaciones en el gen REN (renina): son muy raras. Presentan un fenotipo con hiperuricemia, insuficiencia renal, tensión arterial normal o baja e hiperpotasemia leve.
3. Mutaciones en el gen MUC1 (mucina 1): descritas en muy pocas familias, cursan con progresión a insuficiencia renal, sin hiperuricemia ni gota.
4. Mutaciones en el gen HNF1β (factor nuclear hepático 1b/TFC2): solo se incluyen como NTAD las formas clínicas en las que la fibrosis tubulointersticial es la manifestación principal. Puede asociarse a: diabetes MODY tipo 5, a la presencia de malformaciones renales y genitourinarias (riñón único o riñón en herradura), hiperuricemia, hipomagnesemia, atrofia pancreática y alteraciones de la función hepática(2,15).
5. Mutación no especificada.
Enfermedades quísticas renales no hereditarias
Displasia renal multiquística
La displasia renal multiquística es la forma más grave de displasia renal. Constituye una de las malformaciones quísticas más frecuentes en la infancia y la segunda causa de masa abdominal en el periodo perinatal tras la hidronefrosis(1).
Se caracteriza por la presencia de múltiples quistes, no comunicantes entre sí y rodeados de tejido conectivo fibroso, que sustituyen al parénquima renal. El uréter, con frecuencia es atrésico o está ausente. Puede afectar a ambos riñones, en cuyo caso sería incompatible con la vida o daría lugar a ERT en la etapa neonatal, pero lo más habitual es la forma unilateral, con ligero predominio en el riñón izquierdo(16).
Su incidencia es de 1 de cada 3.640-4.300 recién nacidos vivos(1,16,17), con predominio en el sexo masculino. Con frecuencia, se asocia a alteraciones genitourinarias en el riñón contralateral. Lo más frecuente es el reflujo vesicoureteral (5-43%), siendo este habitualmente de bajo grado (grados I y II). Puede asociarse también a: uropatía obstructiva (estenosis de la unión pieloureteral [7-15%], estenosis ureterovesical [6%]) y anomalías genitales ipsilaterales (15%). Se ha descrito también la presencia concomitante de alteraciones extrarrenales (cardiacas, gastrointestinales, neurológicas y musculoesqueléticas)(16).
Su diagnóstico es ecográfico. En la mayoría de los casos, se detecta en las ecografías prenatales, como un conjunto de quistes de diferentes tamaños, no comunicantes entre sí, rodeados por escaso estroma hiperecogénico. El principal diagnóstico diferencial debe establecerse con las displasias secundarias a obstrucción de la vía urinaria(17).
El riñón contralateral, si es normal, presentará una hipertrofia compensadora incluso al nacimiento. Si esta hipertrofia compensadora no se produce, debe sospecharse la existencia de alguna anomalía asociada en este riñón.
Tradicionalmente, se recomendaba la nefrectomía del riñón multiquístico unilateral, con el objetivo de prevenir posibles complicaciones, como la HTA o la degeneración maligna a tumor de Wilms. Sin embargo, la tendencia natural del riñón multiquístico es a la involución, y estudios más recientes y con mayor tamaño muestral han demostrado que la incidencia de estas complicaciones es muy baja, prácticamente similar a la de la población general; por lo que, actualmente, no se recomienda la nefrectomía(16,17).
Por otro lado, al tratarse de pacientes con un riñón único funcionante, existe el riesgo de sobrecarga funcional, que dé lugar a un estado permanente de hiperfiltración, con proteinuria y deterioro de la función renal a largo plazo. Por ello, se recomienda un seguimiento prolongado, que incluya: controles ecográficos periódicos para confirmar el adecuado crecimiento del riñón contralateral, así como la ausencia de alteraciones en el mismo, sugestivas de reflujo vesicoureteral o de otras anomalías renales; análisis de orina para evaluar hematuria y proteinuria; y control de la función renal y de la presión arterial(1,16,17).
Quiste renal simple y quiste renal complejo o multilocular
Los quistes renales simples suelen ser benignos y asintomáticos. La clasificación de Bosniak nos permite diferenciarlos de los quistes complejos o multiloculares, con riesgo de malignidad.
Los quistes renales simples son poco frecuentes en la edad pediátrica, con una prevalencia de 0,22-2%, aunque su incidencia aumenta con la edad, siendo comunes en la edad adulta(18). Suelen ser benignos y asintomáticos. Las complicaciones más frecuentes son: la hemorragia, la infección o la presencia de un quiste de gran tamaño que pueda comprimir el parénquima renal.
El principal reto diagnóstico es diferenciar el quiste simple del quiste renal complejo o multilocular, que asocia riesgo de malignidad. La clasificación de Bosniak, basada en las imágenes obtenidas mediante tomografía computerizada (TC), es el método de referencia en adultos para el diagnóstico y manejo de los quistes renales (Tabla V)(19).
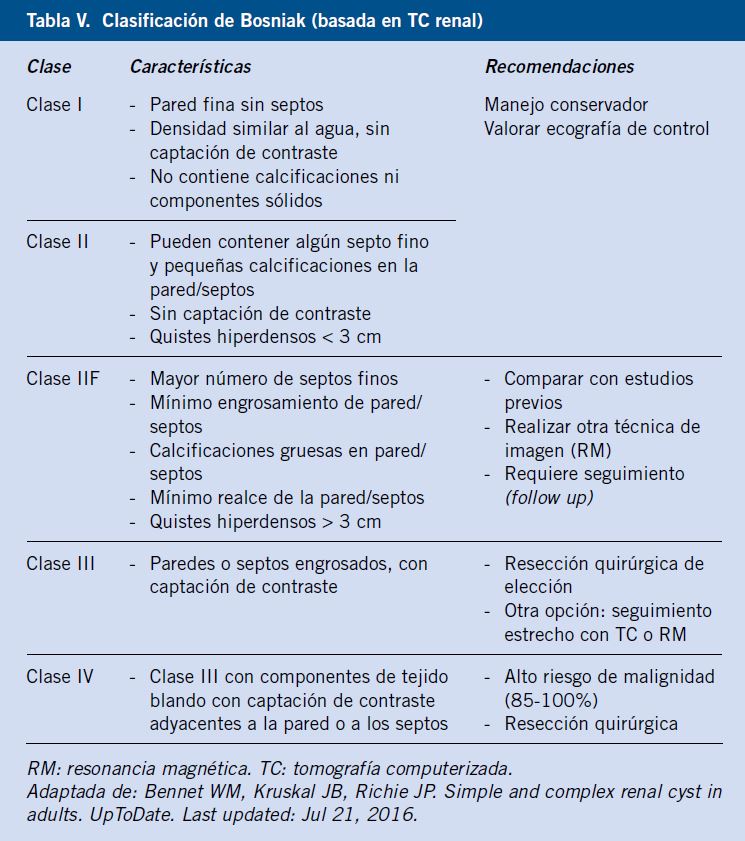
Sin embargo, la degeneración maligna de los quistes renales es una complicación muy poco frecuente en la población pediátrica(18). Se ha propuesto una clasificación de Bosniak modificada (Tabla VI), basada en hallazgos ecográficos (técnica sencilla, económica, no ionizante) para el manejo diagnóstico inicial en niños, evitando el uso sistemático del TC(18).
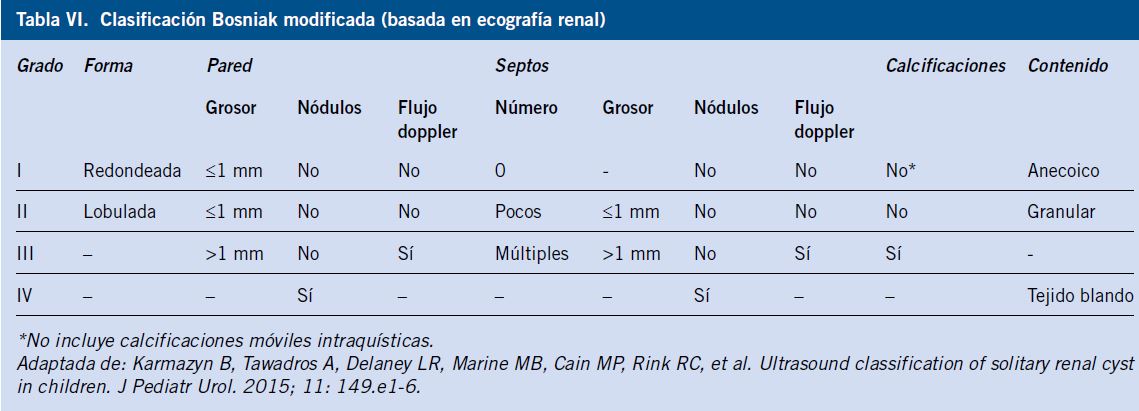
Sus autores, recomiendan un manejo conservador del quiste simple (grados I y II) en la población pediátrica, con seguimiento ecográfico, reservándose la realización de otras técnicas de imagen (TC, RM…) y la cirugía para los quistes complejos (grados III y IV) o los quistes simples complicados(1,18).
Riñón glomeruloquístico
Se caracteriza por la dilatación quística de los glomérulos renales, pudiendo estar asociado a diferentes etiologías que los clasifican en 5 subgrupos.
Los quistes glomerulares se definen como una dilatación del espacio de Bowman 2 o 3 veces superior al límite normal. Hablaremos de riñón glomeruloquístico cuando, al menos, un 5% de sus glomérulos sean quísticos(1,2,20). Lennerz clasificó el riñón glomeruloquístico (RGQ) en 5 subtipos(20):
• Tipo I. RGQ asociado a poliquistosis renal: más frecuentemente asociado a la PQRAD de debut precoz en la infancia.
• Tipo II. RGQ hereditario (o enfermedad glomeruloquística renal, EGQR): sigue una herencia autosómica dominante y engloba 3 formas: una EGQR debida a mutaciones en el gen UMOD, una EGQR familiar hipoplásica secundaria a mutaciones en HNF-1bβ y una tercera debida a mutaciones en genes diferentes a los mencionados previamente.
• Tipo III. RGQ sindrómico: RGQ asociado a un síndrome clínico conocido, que no incluya displasia renal, siendo el más frecuente la esclerosis tuberosa.
• Tipo IV. RGQ obstructivo:asociado a displasia renal o a obstrucción del tracto urinario sin displasia renal, sin evidencia de alteraciones hereditarias.
• Tipo V. RGQ esporádico: recoge aquellos casos que no se pueden incluir en los grupos anteriores, siendo la etiología isquémica (esclerosis sistémica, síndrome hemolítico urémico) y la secundaria a fármacos (litio), las más frecuentes.
El tratamiento será el sintomático de la ERC y etiológico en los casos asociados a obstrucción o inducidos por fármacos(1).
Bibliografía
Los asteriscos destacan los artículos de interés a juicio del autor.
1.** Iceta Lizarraga A, Barajas de Frutos D. Enfermedades quísticas renales. Protoc diagn ter pediatr. 2014; 1: 191-206.
2.*** Loftus H, Ong AC. Cystic kidney diseases: many ways to form a cyst. Pediatr Nephrol. 2013; 28: 33-49.
3.** Sweeney WE Jr, Avner ED. Diagnosis and management of childhood polycystic kidney disease. Pediatr Nephrol. 2011; 26: 675-92.
4. Costa T, Pereira E. Enfermedad renal poliquística autosómica recesiva. En: García Nieto V, Santos Rodríguez F, Rodríguez-Iturbe B, ed. Nefrología pediátrica. 2ª ed. Madrid: Grupo Aula Médica S.L.; 2006. p. 893-900.
5. Guay-Woodford LM, Bissler JJ, Braun MC, Bockenhauer D, Cadnapaphornchai MA, Dell KM, et al. Consensus expert recommendations for the diagnosis and management of autosomal recessive polycystic kidney disease: report of an international conference. J Pediatr. 2014; 165: 611-7.
6. Ariceta Iraola G, Lens Neo XM. Poliquistosis renal autosómica dominante. En: García Nieto V, Santos Rodríguez F, Rodríguez-Iturbe B, ed. Nefrología pediátrica. 2ª ed. Madrid: Grupo Aula Médica S.L.; 2006. p. 883-92.
7. Ong AC, Devuyst O, Knebelmann B, Walz G, ERA-EDTA Working group for inherited kidney diseases. Autosomal dominant polycystic kidney disease: the changing face of clinical management. Lancet. 2015; 385: 1993-2002.
8. Laass MW, Spiegel M, Jauch A, Hahn G, Rupprecht E, Vogelberg C, et al. Tuberous sclerosis and polycystic kidney disease in a 3-month-old infant. Pediatr Nephrol. 2004; 19: 602-608.
9. Torra R. Apuntes sobre el uso del Tolvaptán para la poliquistosis renal autosómica dominante. Resumen de las recomendaciones del grupo de trabajo de enfermedades renales hereditarias de la EDTA y ERBP.
10. Ficha técnica de Jinarc® (Tolvaptán).
11. Safety, Pharmacokinetics, Tolerability and Efficacy of Tolvaptan in Children and Adolescents with ADPKD (Autosomal Dominant Polycystic Kidney Disease). Available from: https://clinicaltrials.gov. Identifier NCT02964273.
12. Fernández Escribano A, Aparicio C, Izquierdo E. Complejo nefronoptisis. Enfermedad quística medular. En: García Nieto V, Santos Rodríguez F, Rodríguez-Iturbe B, ed. Nefrología pediátrica. 2ª ed. Madrid: Grupo Aula Médica S.L.; 2006. p.901-8.
13. Wolf MTF, Hildebrandt F. Nephronophthisis. Pediatr Nephrol. 2011; 26: 181-94.
14. Stokman M, Lilien M, Knoers N. Nephronophthisis. En: Pagon RA, Adam MP, Ardinger HH, et al, ed. GeneReviews® [Internet]. Seatle (WA): University of Washington, Seatle; 1993-2017.
15. Ayasreh Fierro N, Miquel Rodríguez R, Matamala Gastón A, Ars Criach E, Torra Balcells R. Revisión de la nefropatía tubulointersticial autosómica dominante. Nefrología. 2017; 37: 235-43.
16. Valenciano Fuente B, del Campo Casanelles M, Martí Herrero M. Displasia renal multiquística. Afectación nefrourológica en los síndromes polimalformativos. En: García Nieto V, Santos Rodríguez F, Rodríguez-Iturbe B, ed. Nefrología pediátrica. 2ª ed. Madrid: Grupo Aula Médica S.L.; 2006. p.901-8.
17. Hains DS, Bates CM, Ingraham S, Schwaderer AL. Management and etiology of the unilateral multicystic dysplastic kidney: a review. Pediatr Nephrol. 2009; 24: 233-41.
18. Karmazyn B, Tawadros A, Delaney LR, Marine MB, Cain MP, Rink RC, et al. Ultrasound classification of solitary renal cyst in children. J Pediatr Urol. 2015; 11: 149.e1-6.
19. Bennet WM, Kruskal JB, Richie JP. Simple and complex renal cyst in adults. UpToDate. Last updated: Jul 21, 2016.
20. Lennerz JK, Spence DC, Iskandar SS, Dehner LP, Liapis H. Glomerulocystic kidney: one hundred-year perspective. Arch Pathol Lab Med. 2010; 134: 583-605.
Bibliografía recomendada
– Loftus H, Ong AC. Cystic kidney diseases: many ways to form a cyst. Pediatr Nephrol. 2013; 28: 33-49.
Artículo de revisión de las principales patologías quísticas renales, que también analiza las teorías referentes a su patogenia, como la alteración de la función ciliar.
– Iceta Lizarraga A, Barajas de Frutos D. Enfermedades quísticas renales. Protoc diagn ter pediatr. 2014; 1: 191-206.
Protocolo de la asociación española de nefrología pediátrica, actualizado recientemente, que revisa de forma práctica y breve el manejo de las enfermedades quísticas renales.
| Caso clínico |
|
|
