Hepatomegalia
Introducción
La hepatomegalia puede ser debida a una enfermedad hepática o a una enfermedad generalizada.
La hepatomegalia es un signo físico detectado con relativa frecuencia en la consulta del pediatra que no debe menospreciarse; puesto que, puede ser la manifestación de una hepatopatía o de un trastorno sistémico con expresión hepática.
Algunas situaciones clínicas que pueden condicionar la palpación de una “falsa hepatomegalia” son las anomalías morfológicas de la caja torácica (el pectus excavatum, el tórax estrecho por constitución asténica) o los procesos respiratorios que cursan con descenso del diafragma, como el broncoespasmo o el neumotórax. Una variante normal del lóbulo hepático derecho, llamado “lóbulo de Riedel”, puede extenderse por debajo del reborde costal e interpretarse como hepatomegalia; las personas con esta variante están asintomáticas y no presentan signos clínicos ni analíticos de hepatopatía. Otra consideración importante es que la presencia de algunas masas abdominales (quiste de colédoco, masa retroperitoneal, abscesos perihepáticos, vesícula biliar distendida) pueden ser confundidas con hepatomegalia en la palpación.
Etiopatogenia
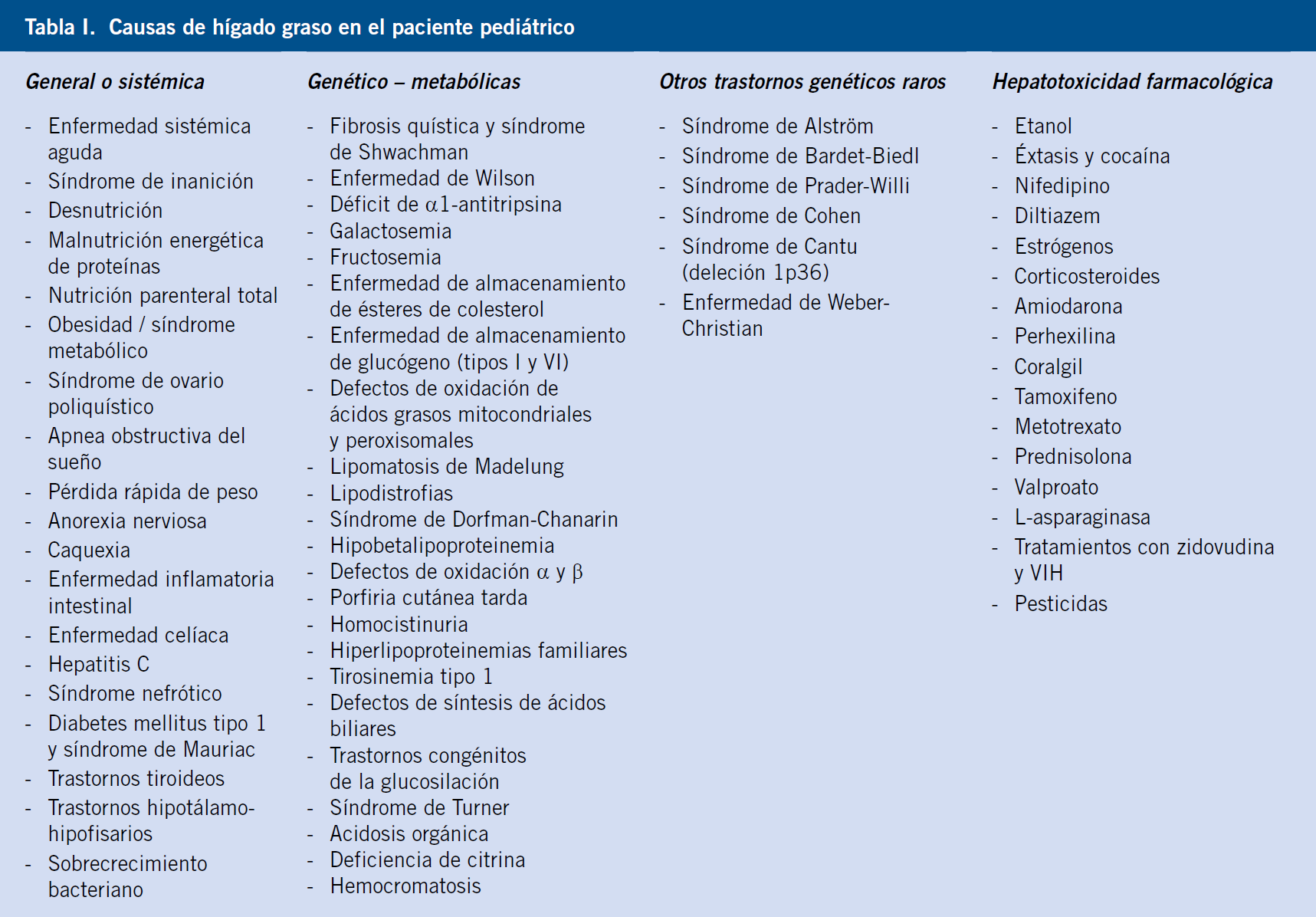
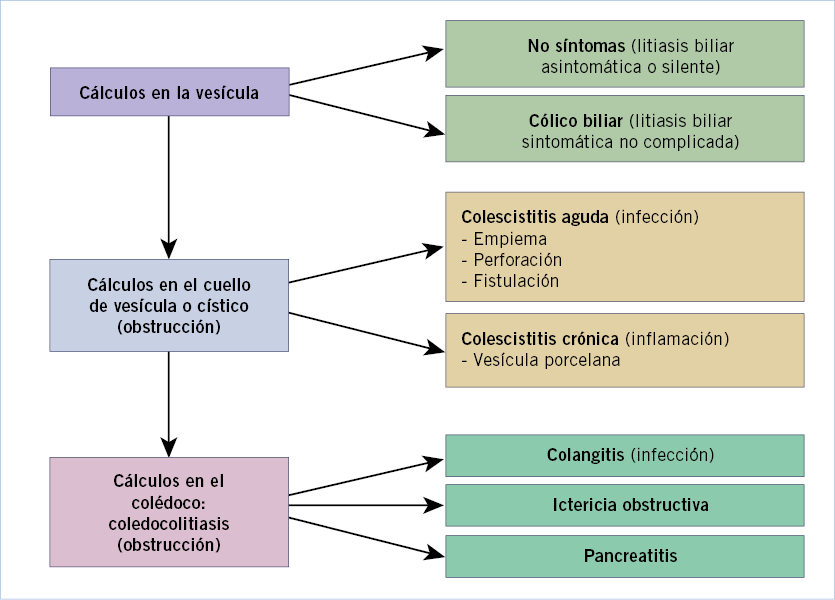
La hepatomegalia puede ser originada por cinco mecanismos diferentes: inflamación, depósito excesivo, infiltración, congestión y obstrucción.
1. Inflamación. Las infecciones, los tóxicos, las radiaciones, enfermedades autoinmunes y la hiperplasia de células de Kupffer inducen hepatomegalia mediada por mecanismo inflamatorio.
2. Depósito. Las sustancias que pueden depositarse en exceso en el hígado originando hepatomegalia son: glucógeno, lípidos, grasa, metales y proteínas anormales.
3. Infiltración. La infiltración es el mecanismo de la hepatomegalia en el caso de tumores, quistes parasitarios y hematopoyesis extramedular. Las células tumorales pueden tener su origen en tumores primarios hepáticos benignos o malignos o en tumores extrahepáticos (metástasis). Las células que infiltran el hígado en el caso de hematopoyesis extramedular y en los síndromes hemofagocíticos son células sanguíneas.
4. Congestión vascular. La obstrucción al drenaje venoso entre el hígado y la aurícula derecha origina hepatomegalia. La obstrucción puede localizarse a nivel intrahepático o extrahepático.
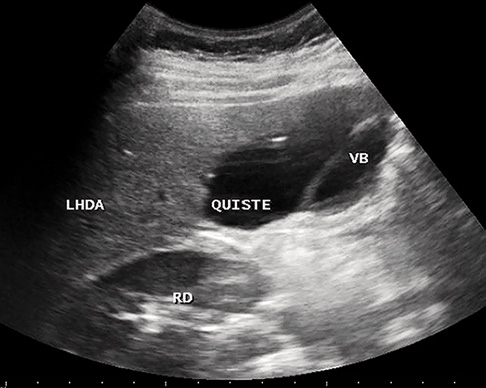
5. Obstrucción biliar. La obstrucción al flujo biliar es el mecanismo de la hepatomegalia en la atresia biliar, quistes de colédoco, colelitiasis y tumores de localización hepática, biliar, pancreática y duodenal (Tabla I).

Actitud ante un paciente pediátrico con hepatomegalia
La anamnesis y la exploración física, junto con las pruebas complementarias básicas (hemograma, coagulación, función hepática, gasometría, sedimento urinario y ecografía), permiten orientar al paciente pediátrico con hepatomegalia y seleccionar el resto de pruebas complementarias a realizar.
El proceso diagnóstico ante una hepatomegalia detectada en un paciente pediátrico incluye una minuciosa anamnesis, exploración física completa y pruebas complementarias de primer nivel. Esta orientación inicial nos debe permitir distinguir si nos enfrentamos a un proceso agudo o crónico y si el proceso se puede manejar ambulatoriamente o requiere atención hospitalaria urgente. Asimismo, nos debe ayudar a discernir si el paciente presenta una hepatopatía o una enfermedad sistémica que cursa con hepatomegalia, con el fin de completar el estudio etiológico e iniciar el tratamiento adecuado de la forma más precoz posible(1).
Anamnesis (Tabla II)

Antecedentes personales
• Embarazo: datos compatibles con infección connatal, adicción materna a drogas, crecimiento intrauterino retrasado (en infección connatal, en algunas enfermedades metabólicas).
• Período neonatal: peso de recién nacido (bajo peso en síndrome de Alagille), canalización de vena umbilical (posible hipertensión portal por cavernomatosis portal), ictericia neonatal, historia de incompatibilidad ABO o Rh (hematopoyesis extramedular por hemólisis), nutrición parenteral prolongada, retraso en la eliminación del meconio (fibrosis quística). Resultado de pruebas metabólicas.
• Desarrollo psicomotor (importante en metabolopatías). Antecedente de convulsiones.
• Curva ponderoestatural. Introducción y tolerancia de alimentación complementaria, aversión a la fruta o dulce (en fructosemia). Características de deposiciones, vómitos.
• Prurito (en colestasis).
• Sangrado (epistaxis, sangrado digestivo, hematomas sin traumatismo), como dato de insuficiencia hepatocelular o pancitopenia por proceso maligno.
• Calendario vacunal. Antecedente transfusional, de tatuajes o piercing, uso de drogas por vía parenteral. Contacto con animales. Viajes (investigación de transmisión de infecciones).
• Infecciones de repetición. Broncoespasmos (fibrosis quística).
• Antecedente de inmunodeficiencia, enfermedad inflamatoria intestinal (asociación de ambas patologías con colangitis esclerosante). Artralgias, exantemas (colagenosis). Antecedente de cardiopatía.
• Fármacos relacionados con hepatomegalia: antiinflamatorios no esteroideos, isoniacida, propiltiouracilo y sulfonamidas.
Antecedentes familiares
• Consanguinidad, historia de abortos de repetición o mortinatos, enfermedades neurodegenerativas (apoyaría enfermedad metabólica).
• Hepatitis víricas (B, C), anemia hemolítica, enfermedades autoinmunes, hepatopatía crónica, hipercolesterolemia, enfermedad renal.
Enfermedad actual
La primera cuestión a plantearse es si se trata de una hepatomegalia asintomática o existen síntomas acompañantes. La anamnesis se debe orientar según la edad del paciente.
Interrogar sobre datos de infección: fiebre, exantemas, aparición de adenopatías, astenia, odinofagia. Contacto con animales, vivienda en zona endémica de leishmaniasis. Convivencia con personas con hepatitis.
Investigar síntomas de enfermedad hepática: colestasis (coluria, acolia, ictericia, prurito) o insuficiencia hepática (diátesis hemorrágica, hipoglucemia, encefalopatía: recordar que la clínica de encefalopatía en lactantes puede ser inespecífica, los padres lo encuentran “raro, irritable”). Valorar clínica de hepatitis aguda: anorexia, astenia, vómitos, febrícula, coluria o dolor abdominal en hipocondrio derecho.
Interrogar sobre síntomas tumorales: malestar, astenia, palidez, distensión abdominal, fiebre prolongada, pérdida de peso o cambio de ritmo intestinal.
Investigación de errores congénitos del metabolismo, importante sobre todo en recién nacidos y lactantes. En el recién nacido, la presentación clínica suele ser grave y puede ir precedida por un período de normalidad de horas o días de duración. La enfermedad puede presentarse como un cuadro inespecífico de: vómitos, letargia, rechazo de las tomas, mala succión, hipotonía, apneas, dificultad respiratoria o “aspecto séptico”. En lactantes, algunas metabolopatías se presentan con manifestaciones hepáticas y extrahepáticas de carácter grave, precipitadas por infecciones intercurrentes, vómitos y ayunos prolongados. En el caso de la fructosemia, la clínica aparece tras la introducción de la fructosa. En niños mayores, es importante preguntar por clínica de hipoglucemia (palidez, sudoración) ante ayunos prolongados (en glucogenosis).
Exploración física (Tabla II)
La exploración física minuciosa es de gran utilidad en el paciente con hepatomegalia. No debe centrarse solo en la exploración abdominal; ya que, en muchas ocasiones, la valoración global y del resto de órganos es lo que nos permite orientar el diagnóstico.
• Peso, talla, perímetro cefálico, percentiles. Estado de nutrición, hábito malabsortivo. El retraso ponderoestatural sugiere enfermedad crónica. Fenotipo (cromosomopatías, enfermedades metabólicas, síndrome de Alagille…). Acropaquias, edemas periféricos. Fetor hepático por hiperamoniemia. Olor especial de la orina: olor a jarabe de arce (enfermedad de la orina de jarabe de arce), olor a pies sudados (acidemias orgánicas), olor a repollo cocido (tirosinemia), u olor a ratón (fenilcetonuria).
• Piel y mucosas: exantemas (infección viral, colagenosis), palidez (anemia, tumores), hematomas o petequias (coagulopatía o pancitopenia), ictericia (conjuntival y/o cutánea en situaciones de hemólisis o alteración hepática), xantomas (en colestasis y otras causas de hipercolesterolemia), lesiones de rascado (en colestasis), angiomas (hemangioma).
• Adenopatías (mononucleosis infecciosa, otras infecciones, tumores).
• Dificultad respiratoria (falsa hepatomegalia, insuficiencia cardíaca).
• Auscultación cardiopulmonar (cardiopatía, pericarditis, taponamiento, soplo por anemia).
• Abdomen: ascitis (síndrome de Budd-Chiari y otras hepatopatías), circulación colateral (en hipertensión portal), palpación de masas abdominales, esplenomegalia (llamativa en infecciones como kala-azar, enfermedades de depósito e infiltración por células malignas).
En la valoración de la hepatomegalia, se debe incluir el tamaño del hígado expresado en centímetros, la homogeneidad, las características regulares o irregulares del borde hepático, la palpación o no del lóbulo izquierdo y la consistencia hepática (en general, consistencia blanda orienta a enfermedades de depósito o hígado de éstasis, y la consistencia aumentada a cirrosis o fibrosis hepática congénita). El dolor a la palpación hepática aparece solo en las hepatomegalias de instauración brusca y se produce por distensión de la cápsula de Glisson.
• Datos de hepatopatía crónica: spiders, lesiones de rascado, eritema palmar, ictericia, ascitis, circulación colateral abdominal, acropaquias…
Pruebas complementarias de primer nivel
Las pruebas complementarias que se deben realizar a todos los pacientes en los que se detecta hepatomegalia son: análisis de sangre y orina y ecografía doppler abdominal.
• Analítica de sangre:
- Hemograma con recuento diferencial y frotis de sangre periférica (buscando blastos, linfocitos estimulados). Reticulocitos (hemólisis). Velocidad de sedimentación.
- Coagulación: actividad de protrombina, tiempo de cefalina, fibrinógeno. Plaquetas.
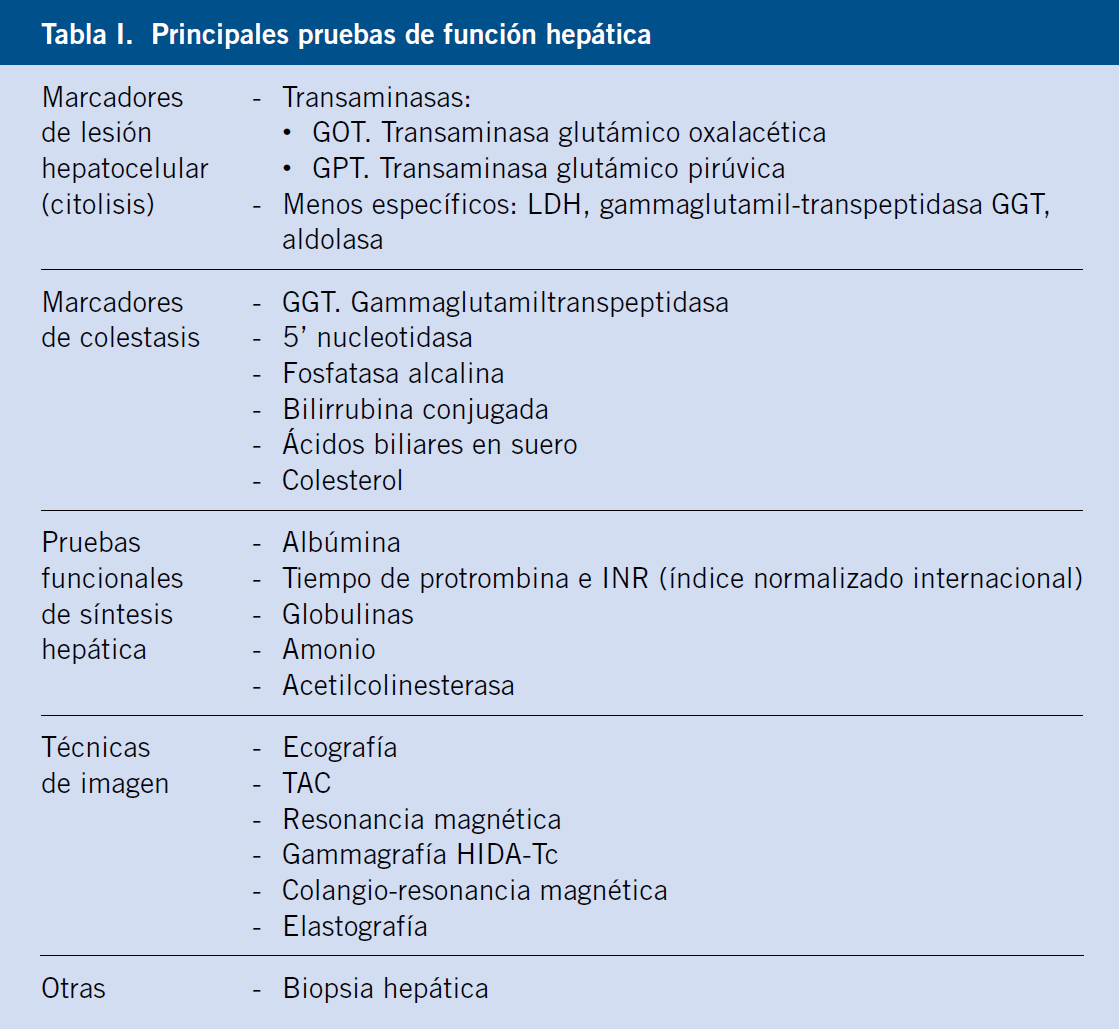
- Bioquímica: función renal (creatinina, urea, iones), gasometría, glucemia, colesterol, LDH (hemólisis), bilirrubina indirecta (hemólisis), función hepática (datos de necrosis: ALT, AST; datos de colestasis: GGT, bilirrubina total y fraccionada, fosfatasa alcalina; y datos de síntesis: actividad de protrombina, glucemia, colesterol, colinesterasa, proteínas totales y albúmina), triglicéridos y enzimas musculares (CPK, aldolasa).
• Análisis de orina: sedimento y urocultivo (en recién nacido y lactante).
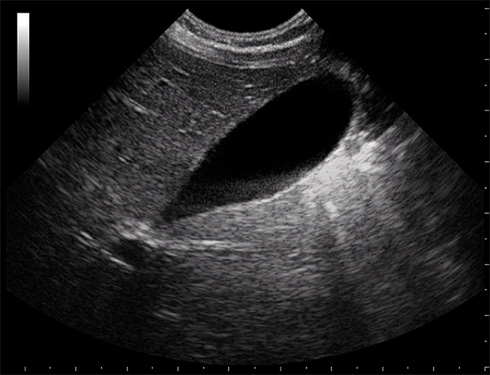
• Ecografía doppler abdominal: es la técnica de imagen de elección en la valoración inicial de la hepatomegalia. Determina el tamaño del hígado, la homogeneidad del parénquima, identifica masas o quistes, cálculos y barro biliar. La técnica doppler permite valorar la permeabilidad de los vasos, el calibre y flujo portal, así como la existencia de circulación colateral. No es una buena prueba para descartar trombosis de las suprahepáticas.
Consideraciones importantes iniciales ante una hepatomegalia
• Si el paciente presenta un proceso agudo infeccioso que cursa con broncoespasmo y hepatomegalia, sin otros datos de enfermedad grave, es conveniente repetir la exploración unos días después para confirmar la hepatomegalia y realizar pruebas complementarias si son necesarias.
• Los pacientes que presenten signos clínicos de gravedad o compatibles con proceso tumoral y los recién nacidos en los que la hepatomegalia se acompañe de colestasis, deben ser remitidos a un centro hospitalario, con el fin de agilizar el diagnóstico y el tratamiento. Los pacientes con hepatomegalia y analítica o sospecha clínica de fallo hepático deben ser remitidos de forma inmediata a un centro hospitalario con equipo de trasplante hepático.
• En todos los casos de hepatomegalia, se deben realizar los estudios de primer nivel de la forma más rápida posible con objeto de orientar al paciente y seleccionar los estudios posteriores. Así: la ecografía identificará tumores abdominales; el hemograma sugerirá procesos infecciosos o tumorales; la coagulación discriminará situaciones de fallo hepático; la bioquímica hepática orientará a cuadros con predominio de colestasis o con predominio de necrosis; la gasometría, cuerpos cetónicos en orina, glucemia y amonio permitirán iniciar el estudio de metabolopatías, etc.
• El planteamiento es diferente según la edad del paciente. Simplificando, abordaremos el diagnóstico en dos grupos de edad: recién nacidos y lactantes hasta el año y en los niños mayores de un año.
Otras pruebas complementarias
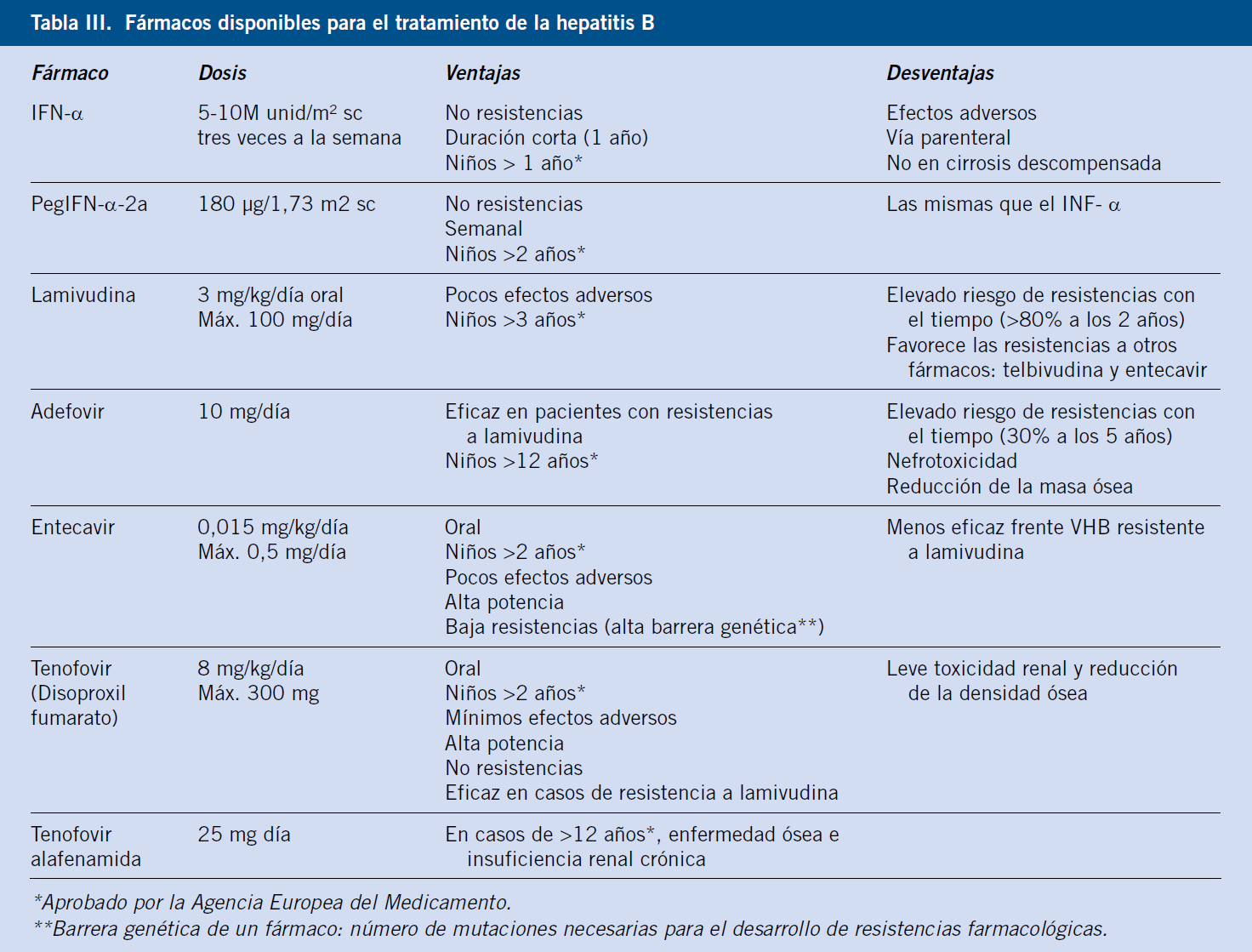
En función de la orientación diagnóstica inicial, estarán indicadas otras pruebas complementarias más específicas (Tabla III).

Analítica (Tabla III)
Pruebas de imagen
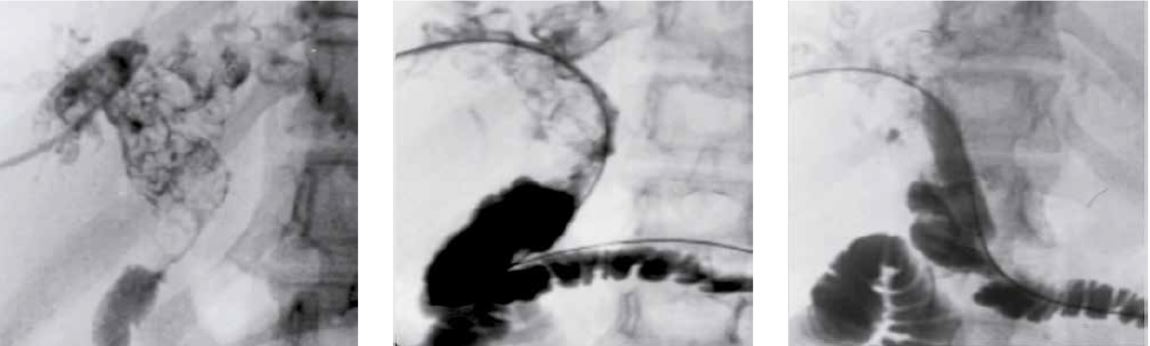
La prueba de imagen de elección en la valoración inicial de la hepatomegalia es la ecografía doppler (ver pruebas complementarias de primer nivel). Sin embargo, la tomografía axial computerizada (TAC) y la resonancia magnética nuclear pueden ser superiores a la ecografía para detectar o definir lesiones focales pequeñas, como: tumores, quistes y abscesos. En patología tumoral, es necesario realizar TAC tóraco-abdominal para valorar la extensión. La angioRMN y angioTACpermiten definir mejor que la ecografía la morfología y la permeabilidad vascular (más útil en la sospecha de síndrome de Budd-Chiari). En sospecha de patología biliar, la colangioRMN es de gran ayuda. Los avances en radiología intervencionista han permitido el diagnóstico y el tratamiento de algunas patologías biliares (mediante colangiografía transparietohepática, que valora el árbol biliar intra y extrahepático) y vasculares (cavografía).
Anatomía patológica
La biopsia hepática por punción, nos permite estudiar la histología del parénquima hepático, informándonos del grado de lesión hepática y de la etiología del proceso responsable de la hepatomegalia. La aplicación de tinciones especiales permite detectar depósitos anómalos. El estudio enzimático en tejido hepático puede requerir biopsia en cuña. La biopsia de médula ósea y punción-aspiración está indicada ante la sospecha de proceso hematológico maligno, de leishmaniasis o de síndrome hemofagocítico, y puede orientar al diagnóstico de algunas enfermedades de depósito (Gaucher, Niemann-Pick). La biopsia muscular ante la sospecha de enfermedad mitocondrial. La biopsia de pielen la enfermedad de Gaucher, enfermedad de Niemann-Pick, enfermedad de Wolman, depósito de ésteres de colesterol, Zellweger y trastornos de la glicosilación. La biopsia de glándula salival ante la sospecha de hemocromatosis neonatal.
Exploración oftalmológica
La exploración oftalmológica puede ser de ayuda en el diagnóstico de enfermedad de Wilson (anillo de Kayser-Fleischer, cataratas), de galactosemia (cataratas), de algunas enfermedades por depósito de lípidos (mancha rojo cereza), de infección connatal (coriorretinitis) o de síndrome de Alagille (embriotoxon posterior).
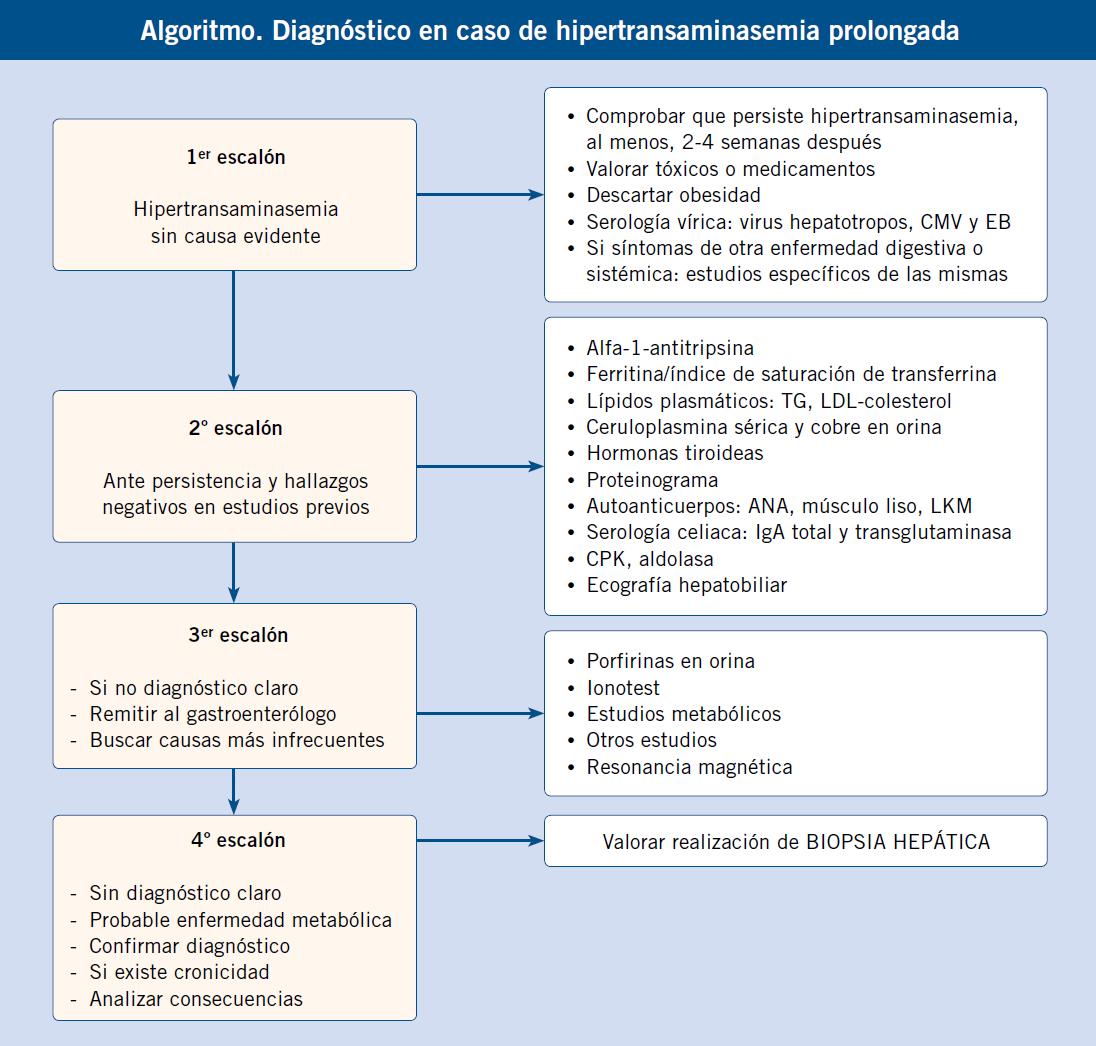
Enfoque diagnóstico de la hepatomegalia en recién nacidos y lactantes. (Ver algoritmo 1 al final del artículo)
En los recién nacidos, la anamnesis, exploración física y pruebas complementarias deben ir dirigidas a detectar enfermedades graves que sean susceptibles de recibir tratamiento precoz que modifique favorablemente su pronóstico.
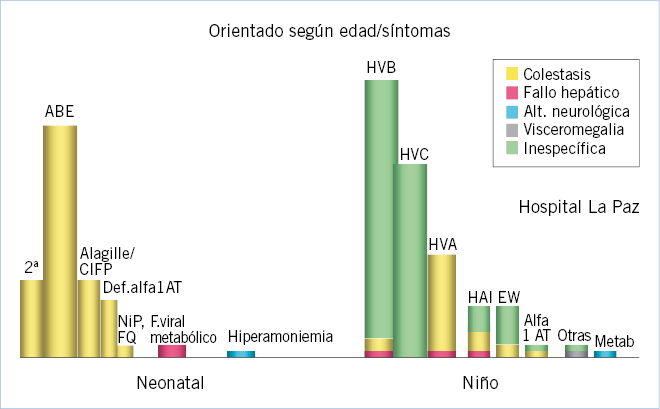
En la valoración de la hepatomegalia neonatal, se deben considerar los signos acompañantes. Si la hepatomegalia es el dato dominante en la exploración, orienta a tumores de origen hepático y extrahepático (los hepáticos más frecuentes a esta edad son los hemangiomas y los hepatoblastomas) y a enfermedades metabólicas de depósito (glucogenosis tipo I, III, cursan sin esplenomegalia; desordenes congénitos de la glicosilación, Gaucher, enfermedades peroxisomales). En muchas ocasiones, la hepatomegalia se acompaña de colestasis(2), predominando esta última en la enfermedad. El hígado neonatal y del lactante es inmaduro; de forma que, desarrolla colestasis como expresión de enfermedades hepáticas y sistémicas graves. Entre los cuadros de colestasis neonatal con hepatomegalia, es necesario discernir dos grandes grupos de patologías, las que cursan con fallo hepático y las que no lo hacen. Las que cursan con fallo hepático (convirtiéndose en la manifestación guía) son: galactosemia, fructosemia, tirosinemia, hemocromatosis, enfermedad de Wolman, TORCH y VHB vertical (>45 días). Entre las que no cursan con fallo hepático, lo más importante es descartar la atresia biliar extrahepática, ya que su pronóstico depende de la corrección quirúrgica temprana (antes de los 2 meses de edad). Se sospecha por ictericia persistente con acolia (mejor ver las deposiciones). Otras causas de colestasis sin fallo hepático, en el período neonatal y de lactante (ver capítulo correspondiente) son: el déficit de alfa-1-antitripsina, síndrome de Alagille, colestasis intrahepática familiar progresiva, fibrosis quística, enfermedad de Niemann-Pick A y C, enfermedad de Zellweger, panhipopituitarismo, quiste de colédoco, perforación espontánea de la vía biliar, sepsis, infección urinaria, tóxicos, nutrición parenteral y multifactorial. La presencia de hiperbilirrubinemia indirecta con anemia y hepatoesplenomegalia sugiere hematopoyesis extramedular como causa de la hepatomegalia.
Enfoque diagnóstico de la hepatomegalia en niños mayores de un año. (Ver algoritmo 2 al final del artículo)
La hepatomegalia en este grupo de edad puede ser manifestación de una enfermedad hepática crónica o aguda, de una situación de hematopoyesis extramedular, de tumores de origen hepático o extrahepático, enfermedades de depósito de origen metabólico o infecciones.
Las situaciones de hematopoyesis extramedular cursan con anemia y datos de hemólisis y se acompañan de esplenomegalia. Los tumores más frecuentes son: leucemias, linfomas, hepatoblastoma y hepatocarcinoma. Las enfermedades de depósito a esta edad son: glucogenosis (I, III, VI, IX), enfermedad de Gaucher, enfermedad de Niemann-Pick tipo C y enfermedad por depósito de ésteres de colesterol. La bioquímica hepática ayuda a la orientación del paciente con hepatomegalia y sospecha de hepatopatía aguda y crónica, siendo imprescindible realizar un estudio de coagulación para discriminar las situaciones que se acompañan de fallo hepático. En la bioquímica hepática, pueden predominar los datos de necrosis hepática sobre los de colestasis (elevación de AST y ALT, poca elevación de bilirrubina directa, GGT y fosfatasa alcalina), esto orienta a hepatitis virales, enfermedad de Wilson, hepatitis autoinmunes y tóxicas. Si existe predominio de datos de colestasis sobre la necrosis, la ecografía es de gran utilidad, puesto que permite identificar problemas obstructivos (quistes, malformaciones de la vía biliar, cálculos). Las hepatopatías, como la fibrosis hepática congénita o la poliquistosis hepatorrenal, pueden presentar hepatomegalia con poca alteración funcional. Igualmente, el síndrome de Budd-Chiari suele cursar con poca alteración analítica.
Características clínicas, diagnóstico y tratamiento de algunas enfermedades que cursan con hepatomegalia
A continuación, destacaremos algunas de las enfermedades que pueden cursar con hepatomegalia y señalaremos los datos en la anamnesis, exploración y analítica que orientan a su diagnóstico, así como las pruebas complementarias de segundo nivel que se deben indicar y el tratamiento adecuado. No abordamos en este capítulo las enfermedades infecciosas ni las hepatopatías en las que la hepatomegalia no es el signo guía, puesto que son expuestas en los capítulos restantes de esta monografía.
Enfermedades infecciosas
Infecciones virales: virus Epstein-Barr, citomegalovirus, hepatitis A, B, C y E (ver capítulo de hepatitis agudas).
Protozoos: leishmaniasis visceral o kala-azar (predomina gran esplenomegalia).
Bacterias: fiebre tifoidea (Salmonella typhi)y brucelosis (Brucella melitensis).
Rickettsias: fiebre botonosa mediterránea (Rickettsia conorii).
Otras infecciones: fiebre Q (Coxiella burnetti), tuberculosis, parásitos, candidiasis sistémica, toxoplasma e histoplasmosis. Sepsis, endocarditis. Infección urinaria en el lactante y recién nacido.
Enfermedades metabólicas
Las principales enfermedades metabólicas que cursan con hepatomegalia(3) son: las glucogenosis (fundamentalmente I, III, VI y IX), las enfermedades lisosomales (enfermedades de Gaucher, de Niemann-Pick, de Wolman y por depósito de ésteres de colesterol, mucopolisacaridosis), los defectos de glicosilación de proteínas (CDG) y las enfermedades peroxisomales.
Glucogenosis
Son trastornos del metabolismo del glucógeno y de herencia autosómica recesiva, excepto la tipo IXa (ligada al cromosoma X). El glucógeno es la forma principal de almacenaje de hidratos de carbono, se localiza mayoritariamente en el hígado y músculo; durante los períodos de ayuno y de estrés, el glucagón y la adrenalina estimulan la degradación de glucógeno. Las glucogenosis(4) son trastornos debidos a defectos en los enzimas de degradación o síntesis de glucógeno, lo que condiciona un depósito anormal de glucógeno y/o una deficiente movilización del mismo. La afectación hepática ocurre fundamentalmente en las tipos I, III y IV. Las glucogenosis I, III, VI y IX son las que cursan con hepatomegalia más llamativa.
La glucogenosis tipo Ia (enfermedad de Von Gierke) es debida al defecto de glucosa-6-fosfatasa (cromosoma 17q21), que cataliza el paso final en la glucogenolisis y en la gluconeogénesis en el hígado, riñón e intestino. La presentación clínica típica(5), con hipoglucemia, hepatomegalia de consistencia blanda y acidosis metabólica, ocurre en el período neonatal o de lactante. Los niños presentan hipoglucemias intensas tras breves períodos de ayuno que, generalmente, son bien toleradas; la tolerancia aumenta con la edad. Como consecuencia de la hipoglucemia, pueden producirse convulsiones y secuelas neurológicas. Además de hepatomegalia, cursan con: nefromegalia, nefrocalcinosis, litiasis renal, osteoporosis y con un fenotipo peculiar (cara de muñeca con abdomen prominente, talla baja, obesidad facio-troncular y xantomas); no tienen esplenomegalia ni afectación cardíaca. La hepatomegalia tiende a disminuir con la edad. Los datos analíticos son: hipoglucemia con cetosis leve, acidosis láctica, hipertrigliceridemia muy llamativa, hipercolesterolemia, hiperuricemia, ligera elevación de transaminasas sin aumento de bilirrubina ni alteración de parámetros de síntesis hepática, datos de tubulopatía renal, hipercalciuria, hipocitraturia y alteración en la adhesión y agregación plaquetaria. Es frecuente, evolutivamente, el desarrollo de disfunción renal con glomeruloesclerosis focal y segmentaria e hipertensión arterial. El diagnóstico se basa en el estudio genético o en la demostración del defecto enzimático en la biopsia hepática (precaución por riesgo de sangrado). La histología muestra hepatocitos distendidos con aspecto de célula “vegetal”. El tratamiento consiste en mantener la glucemia en valores normales, evitando períodos prolongados de ayuno; para ello, puede ser necesaria la alimentación por sonda nasogástrica a débito continuo o nocturna. Como aporte de hidratos de carbono, se recomienda dextrinomaltosa y almidón de maíz, evitando la sacarosa, fructosa y galactosa; se debe hacer restricción de grasas y purinas. Están indicados los suplementos de vitamina D y calcio. En el seguimiento de estos pacientes, hay que vigilar la aparición de adenomas hepáticos, con posibilidad de degeneración maligna, mediante controles ecográficos y determinaciones de alfa-fetoproteína. Pueden precisar tratamiento: con alopurinol, inhibidores del enzima convertidor de angiotensina y con hipolipemiantes. La indicación de trasplante hepático es controvertida, porque, aunque corrige el defecto metabólico, normaliza la dieta, mejora la calidad de vida, evita el desarrollo de hepatocarcinoma y permite un catch-upde crecimiento, no parece prevenir o revertir el trastorno renal, que incluso puede agravarse después del trasplante por el efecto nefrotóxico de la medicación inmunosupresora. Las indicaciones de trasplante(6-8) más aceptadas serían la presencia de adenomas múltiples o hepatocarcinoma, aunque debe ser siempre una indicación individualizada; en algunos casos, ha sido realizado trasplante hepático y renal combinado.
En la glucogenosis tipo I b (deficiencia de translocasa de glucosa-6-fosfatasa), la producción de enzima glucosa-6-fosfatasa es normal, pero no puede transportar su sustrato glucosa-6-fosfato. El gen alterado se localiza en el cromosoma 11q23. La presentación clínica, los datos analíticos y las complicaciones son similares a la forma Ia, pero además, presentan neutropenia y alteración en la función de neutrófilos, pudiendo padecer infecciones bacterianas recurrentes y úlceras bucales o intestinales. Está indicado el factor estimulante de colonias. Evolutivamente, pueden presentar un trastorno inflamatorio intestinal semejante a la enfermedad de Crohn. Está descrita una mayor prevalencia de enfermedad autoinmune tiroidea.
La glucogenosis tipo III (enfermedad de Cori o Forbe) se debe al defecto del enzima derramificante amilo-1,6-glucosidasa (cromosoma 1p21), lo que provoca acúmulo de dextrina límite. En el tipo IIIa, el defecto se localiza a nivel hepático, muscular y cardíaco, mientras que en el tipo IIIb, solo a nivel hepático. El tipo IIIa supone el 80% de los casos de glucogenosis tipo III y la presentación es similar a la tipo I, pero puede asociar miocardiopatía hipertrófica obstructiva y afectación muscular, y no cursa con nefromegalia. Las manifestaciones son variables dependiendo de la localización y extensión del defecto enzimático. Los datos analíticos diferentes con la tipo I son los valores normales de láctico, triglicéridos, úrico y la cetonemia intensa en caso de hipoglucemia; pueden presentar elevación de creatinkinasa. El diagnóstico se hace mediante confirmación del defecto enzimático en leucocitos, fibroblastos, eritrocitos, hígado o músculo y mediante estudio genético. El tratamiento es similar al tipo I, pero no es necesaria la restricción de galactosa y fructosa.
La glucogenosis tipo IV (enfermedad de Andersen) por defecto del enzima ramificante (cromosoma 3p12), se presenta inicialmente como disfunción hepática en el lactante y hepatomegalia, progresando rápidamente a cirrosis con hipertensión portal y fallecimiento antes de los cinco años. Es muy poco frecuente y representa el 0,3% de todas las glucogenosis. El acúmulo de glucógeno es generalizado y afecta a hígado, corazón, músculo, piel, intestino, cerebro y sistema nervioso periférico. Diagnóstico mediante determinación enzimática en hígado, músculo, leucocitos o fibroblastos. El trasplante hepático es el único tratamiento disponible, aunque la indicación ha de ser individualizada, puesto que está descrito el desarrollo de complicaciones neurológicas y cardíacas en el seguimiento postrasplante(7,8).
La glucogenosis tipo VI(enfermedad de Hers), por defecto de la fosforilasa hepática (cromosoma 14q 21-22), se presenta en la infancia como hepatomegalia masiva y retraso de crecimiento. Generalmente, están asintomáticos y no presentan hipoglucemia ni acidosis. No tienen afectación cardiológica ni muscular y el pronóstico es bueno con reducción progresiva de la hepatomegalia con la edad.
La glucogenosis tipo IX, por defecto de la kinasa de la fosforilasa hepática, es más frecuente que la VI. Clínica y genéticamente es más heterogénea. La mayoría de los casos son tipo IXa, con herencia ligada al X; debutan entre el año y los 5 años con: hepatomegalia, retraso del crecimiento, hipoglucemia leve y mínima elevación de transaminasas, colesterol y triglicéridos. La hepatomegalia tiende a disminuir con la edad (hepatomegalia benigna). El diagnóstico de los tipos VI y IX se hace mediante determinación enzimática en tejidos afectados, hígado o músculo; es posible la determinación en leucocitos y hematíes, pero la presencia de diferentes isoenzimas puede hacer difícil la interpretación.
Trastornos lisosomales
Los lisosomas son organelas intracelulares que contienen gran número de enzimas, su principal función consiste en la degradación de macromoléculas. Los trastornos de depósito lisosomales son debidos a defectos enzimáticos que ocasionan una acumulación progresiva anómala de sustratos no degradados en el interior de los lisosomas. La mayoría de estas enfermedades tienen herencia autosómica recesiva. El espectro clínico es amplio, los signos que orientan al diagnóstico son: hepatoesplenomegalia (generalmente con predominio de esplenomegalia), retraso en el desarrollo neurológico y rasgos faciales toscos; es frecuente que presenten displasia esquelética. Entre las enfermedades lisosomales que pueden asociar hepatoesplenomegalia, destacan los trastornos por acúmulo de esfingolípidos y lípidos (gangliosidosis GM1, enfermedades de Gaucher, Niemann-Pick, Wolman y por depósito de ésteres de colesterol y Farber), las mucolipidosis y las mucopolisacaridosis.
La enfermedad de Gaucher es el trastorno lisosomal más frecuente, causado por déficit de b-glucosidasa (b-glucocerebrosidasa), con acúmulo de glucosilceramida en los macrófagos (células de Gaucher). Es de herencia autosómica recesiva y el gen afecto se localiza en el cromosoma 1q21. Aunque no existe una clara correlación genotipo-fenotipo, la presencia de, al menos, un alelo N370S parece tener efecto protector frente a la clínica neurológica; si bien, en los últimos años, ha sido descrita una mayor frecuencia de Parkinson y otras alteraciones neurológicas. La mutación L444P en homocigosis, se asocia a formas neuropáticas. Las células de Gaucher se localizan en el hígado, bazo y médula ósea, también en sistema nervioso central, ganglios linfáticos, pulmones y glomérulos; los síntomas derivan del desplazamiento de las células normales por las de Gaucher y del ambiente proinflamatorio promovido por la liberación de citokinas por dichas células. La enfermedad se clasifica en tres tipos de acuerdo a la presencia y severidad de las manifestaciones neurológicas; son comunes a los tres tipos: la hepatoesplenomegalia (la esplenomegalia es un dato constante, la hepatomegalia aparece en el 50%) y la infiltración pulmonar. La afectación ósea ocurre en los tipos 1 y 3. La forma sin afectación neurológica, Gaucher tipo 1, es la más frecuente (94%). La enfermedad de Gaucher tipo 2 o forma neuropática aguda es la menos frecuente, las manifestaciones neurológicas aparecen precozmente en el período de lactante. En la enfermedad de Gaucher tipo 3 o forma neuropática crónica, las manifestaciones neurológicas son variables y de presentación más tardía. Las alteraciones analíticas más destacadas son: pancitopenia, elevación de transaminasas, alteración de la coagulación e hipergammaglobulinemia policlonal. El diagnóstico se basa en la determinación de la actividad enzimática de glucocerebrosidasa en leucocitos o fibroblastos y en el posterior análisis genético. La medición de actividad enzimática en sangre seca sobre papel puede ser utilizada como método de cribado, pero precisa de confirmación por los métodos anteriores. La presencia de células de Gaucher en aspirado de médula ósea y en tejido hepático sugiere esta enfermedad, pero no son específicas. La elevación de la fosfatasa ácida tartrato resistente, la enzima convertidora de angiotensina, CCL-18/PARC (citokina 18) y la quitotriosidasa, que es útil en el diagnóstico y en la monitorización del tratamiento.
Gaucher tipo 1 se presenta a cualquier edad entre la infancia y la edad adulta. Las características son: retraso del crecimiento, hepatoesplenomegalia con gran bazo que condiciona hiperesplenismo y riesgo de rotura, afectación ósea, responsable de gran morbilidad (dolor por infarto, osteoporosis, fracturas patológicas, necrosis avascular), manifestaciones hepáticas (leve alteración funcional, es muy raro el desarrollo de cirrosis o hipertensión portal), y dolor abdominal por infartos esplénicos y hepáticos. Otros problemas son: fallo de la médula ósea (trombopenia y anemia), trombopatía, hipertensión pulmonar y un riesgo aumentado de trastornos linfoproliferativos, mieloma múltiple y hepatocarcinoma. La supervivencia es prolongada. El tratamiento tiene como objetivo el control de los síntomas, la prevención del daño de órganos y la mejoría de la calidad de vida. Existe terapia enzimática sustitutiva intravenosa, inicialmente solo estaba disponible imiglucerasa, pero en los últimos años se han desarrollado velaglucerasa y taliglucerasa, de similar eficacia. La respuesta de la hepatoesplenomegalia y de la afectación medular al tratamiento enzimático sustitutivo(9) es rápida, no así la respuesta del trastorno óseo. No está indicada la esplenectomía, porque empeora los depósitos y acelera el curso de la enfermedad. El tratamiento quirúrgico ortopédico puede ser preciso para el manejo de la afectación ósea, al igual que la analgesia; en caso de crisis óseas, están indicados los esteroides. Los bifosfonatos son utilizados para la osteoporosis. Algunas formas excepcionales con cirrosis pueden requerir trasplante hepático, pero no cura la enfermedad y es necesario continuar el tratamiento enzimático sustitutivo postrasplante. La terapia con miglustat oral, basada en la reducción de sustrato (glucosilceramida) mediante inhibición de su síntesis, no está indicada en niños con enfermedad tipo 1.
La enfermedad de Gaucher tipo 2, de presentación más temprana, puede debutar con hidrops fetal. Los datos principales son hepatoesplenomegalia con afectación neurológica severa que condiciona mortalidad precoz, antes de los 2 años de edad.
Gaucher tipo 3, es de severidad intermedia, se caracteriza por visceromegalia y alteración neurológica con parálisis de la mirada horizontal. Se distinguen 3 subtipos (a, b y c), dependiendo de la preponderancia de los síntomas viscerales o neurológicos. En el tipo 3c, es característica la afectación cardiovascular con calcificación progresiva de grandes arterias y de válvulas mitral y aórtica. El tratamiento enzimático sustitutivo no es eficaz en revertir o prevenir el daño neurológico en los tipos 2 y 3, porque no atraviesa la barrera hematoencefálica; su administración está indicada en el tipo 3 para el control de la afectación visceral. Miglustat no parece tener un impacto significativo sobre la afectación neurológica. El trasplante hepático no es útil y podría estar contraindicado por la afectación neurológica. Actualmente, están en estudio las chaperonas farmacológicas, que tienen como objetivo, incrementar la función residual de las enzimas mutadas.
La enfermedad de Niemann-Pick(10) se produce por depósito de esfingomielina en monocitos-macrófagos (“histiocitos espumosos”) y su herencia es autosómica recesiva.
La enfermedades de Niemann-Pick A y B son debidas al defecto del enzima esfingomielinasa ácida (cromosoma 11p15.4); en el caso de Niemann-Pick A, existe muy poca o ninguna actividad residual enzimática. El depósito ocurre en: bazo, hígado, ganglios linfáticos, médula ósea, riñones y pulmones; en el caso de Niemann-Pick tipo A, se produce también depósito en el sistema nervioso. La enfermedad de Niemann-Pick tipo A debuta en los primeros tres meses de vida, con hepatoesplenomegalia masiva y alteraciones neurológicas precoces (hipotonía, dificultad para alimentarse). Otros datos clínicos son: linfadenopatía, ictericia neonatal prolongada con transformación gigantocelular, vómitos, diarrea, afectación pulmonar con infecciones de repetición y neumonías por aspiración (Rx. con patrón reticular o nodular), mancha rojo cereza en el estudio oftalmológico y fallo para medrar. El retraso psicomotor es evidente a partir de los 6 meses, con rigidez y espasticidad. Fallecen a los 2 años por complicaciones respiratorias.
La enfermedad de Niemann-Pick tipo B tiene una forma de presentación clínica más variable y algunos pacientes son diagnosticados en la edad adulta. Los datos más frecuentes son: hepatoesplenomegalia (esplenomegalia más llamativa que hepatomegalia) que va disminuyendo con la edad y afectación pulmonar con patrón clínico y radiológico similar a la del tipo A, que puede evolucionar a cor pulmonale. Existen datos analíticos de hiperesplenismo y perfil lipídico aterogénico.
El diagnóstico de confirmación es la demostración del déficit enzimático de esfingomielinasa en fibroblastos (biopsia de piel) o en leucocitos y el estudio genético (mutaciones en el gen SPMD1 en el 95% de los casos). La presencia de células espumosas en biopsia de médula ósea o hepática sugiere el diagnóstico, pero no son específicas. No existe tratamiento para el Niemann-Pick A y B, excepto el de soporte; en la actualidad, se está desarrollando terapia sustitutiva enzimática para el tipo B.
La enfermedad de Niemann-Pick tipo C(11) es más frecuente que las anteriores y, clínica, bioquímica y genéticamente diferente a ellas. Es debida en el 95% de los casos a mutaciones en el gen NPC1(cromosoma 18q11-12) y menos frecuentemente a mutaciones en el gen NPC2 (14 q 24.3). La actividad de la esfingomielinasa es normal, pero existe un defecto en el transporte intracelular del colesterol endocitado, lo que condiciona acúmulo de colesterol no esterificado en los lisosomas. Las manifestaciones clínicas son muy heterogéneas, pudiendo presentarse desde la vida intrauterina (ascitis fetal) hasta la edad adulta. Así, son reconocidas la forma prenatal o perinatal, infantil precoz (3 meses-2 años), infantil tardía (2-6 años), juvenil o clásica (6-15 años) y la del adolescente-adulto (>15 años). La enfermedad de Niemann-Pick tipo C es una enfermedad neurovisceral, por tanto, los síntomas posibles son los sistémicos derivados de la afectación visceral (hígado, bazo y, menos frecuentemente, pulmón) y los neurológicos. El componente sistémico puede estar ausente o ser mínimo en un 15% de todos los pacientes, elevándose este porcentaje al 50% en los casos de inicio adulto; cuando está presente, precede al inicio de los síntomas neurológicos, y es bien tolerado, salvo si se manifiesta en el período perinatal. Las manifestaciones en el período perinatal son: colestasis prolongada autolimitada, ascitis y progresivo desarrollo de hepatoesplenomegalia (predomina la esplenomegalia). La mortalidad es elevada en las formas perinatales y viene condicionada por los síntomas sistémicos. Puede haber afectación pulmonar. El trastorno neurológico es progresivo y de inicio no neonatal, cursa con parálisis de la mirada vertical y evolución a oftalmoplejia supranuclear. Es posible la presentación como visceromegalia aislada en lactantes, antes del desarrollo de manifestaciones neurológicas. El diagnóstico se basa en la biopsia de piel, demostrando disminución del nivel de ésteres de colesterol y/o acúmulo de colesterol no esterificado (test de Filipina) en fibroblastos, siendo este último, el método más utilizado. El 70-80% de los pacientes tienen el test de filipina claramente positivo y se denominan: “fenotipo bioquímico clásico”; el porcentaje restante se conoce como: “fenotipo bioquímico variante”, siendo necesario en estos últimos la realización de estudio genético. La presencia de células espumosas e histiocitos “azul marino” en hígado o médula ósea sugiere el diagnóstico. Los biomarcadores: quitotriosidasa, CCL18/PARC y oxiesteroles, suelen estar aumentados. No existe tratamiento específico. El tratamiento precoz con miglustat oral parece conseguir estabilización o mejoría de la sintomatología neurológica, obteniéndose los mejores resultados en los pacientes con formas de inicio tardío. Actualmente, están en investigación otros tratamientos. El fallecimiento suele producirse a partir de la primera década del inicio de la degeneración neurológica, excepto en las formas perinatales, en las que se produce en los primeros meses de vida.
Enfermedad de Wolman y trastorno por depósito de ésteres de colesterol: estas dos enfermedades de herencia autosómica recesiva (gen LIPA en el cromosoma 10q 22,2-22,3), son debidas a deficiencia de lipasa ácida lisosomal, originando acúmulo de ésteres de colesterol y triglicéridos en la mayoría de los tejidos. Se produce un incremento de la síntesis de colesterol, de la producción de lipoproteínas y una regulación positiva de la expresión del gen del receptor LDL. Los cambios son más acusados en la enfermedad de Wolman, porque la deficiencia enzimática es más severa; en el trastorno por depósito de ésteres de colesterol, existe actividad residual enzimática. La enfermedad de Wolman debuta en el período neonatal con: diarrea, vómitos, fallo para medrar y hepatoesplenomegalia. Puede cursar con insuficiencia hepática y el pronóstico es muy malo, con fallecimiento en los primeros meses. En la Rx. de abdomen, es frecuente la detección de calcificaciones suprarrenales. En la enfermedad por depósito de ésteres de colesterol, las manifestaciones son variables. La forma de presentación habitual es la detección casual de hepatomegalia a cualquier edad entre la infancia y la edad adulta. Se acompaña de disfunción hepática y puede haber esplenomegalia, pero son raras la malabsorción, la malnutrición y las calcificaciones adrenales. En la evolución, puede progresar a fibrosis e insuficiencia hepática. Los hallazgos analíticos incluyen: hipercolesterolemia, hipertrigliceridemia y linfocitos vacuolados. La biopsia hepática puede aportar datos del grado de fibrosis y es frecuente la presencia de histiocitos espumosos. El diagnóstico específico de estas dos entidades es la demostración del defecto enzimático de lipasa ácida en fibroblastos (biopsia de piel) o leucocitos. Actualmente, está en marcha un ensayo clínico con terapia enzimática sustitutiva (sebelipasa). El tratamiento clásico en la enfermedad por ésteres de colesterol eran los inhibidores de la enzima HMG CoA reductasa (Simvastatina) y la dieta baja en colesterol; disminuyen el nivel de colesterol, colesterol LDL y triglicéridos, pero no está claro si ejerce efecto beneficioso sobre la disfunción hepática y la hepatoesplenomegalia. El trasplante hepático estaría indicado en casos de afectación severa de la función hepática.
Mucopolisacaridosis: enfermedades con transmisión autosómica recesiva, excepto la mucopolisacaridosis tipo II (Hunter), de transmisión recesiva ligada al cromosoma X. Son debidas a la deficiencia de enzimas necesarias para la degradación de glicosaminglicanos o mucopolisacáridos, lo que condiciona acúmulo de glicosaminglicanos (GAG) dentro de los lisosomas celulares y eliminación de grandes cantidades por la orina; se diferencian distintos subtipos según el defecto enzimático. La sintomatología clínica es debida no solo al depósito de GAG, sino también a la acción de mediadores inflamatorios a nivel celular y extracelular.
Las manifestaciones suelen ser multisistémicas, presentando hepatoesplenomegalia significativa desde edades tempranas, con predominio de la hepatomegalia. Otras manifestaciones frecuentes son: la afectación neurológica, esquelética, cardiovascular, ocular, dérmica, hernias umbilical e inguinales, talla baja y rasgos faciales toscos. La hepatomegalia se observa fundamentalmente en la MPS IH (Hurler), MPS II (Hunter), MPS VI (Maroteaux-Lamy) y MPS VII (Sly). Los tipos MPS IV (Morquio) y MPS III (Sanfilippo) pueden presentar hepatomegalia más leve que en el resto de tipos. La aproximación diagnóstica inicial es la medición de concentración de glicosaminglicanos en orina de 24 horas; la determinación de actividad enzimática en gota de sangre seca es utilizada como método de despistaje. El diagnóstico definitivo debe basarse en el análisis enzimático en leucocitos o cultivo de fibroblastos, recomendándose posteriormente el estudio genético. En los últimos años, se ha producido un cambio significativo en el tratamiento que, instaurado de forma precoz, ha permitido una mejoría significativa de la calidad de vida y la supervivencia. Clásicamente, el tratamiento era paliativo y de las complicaciones. Actualmente, existe tratamiento enzimático sustitutivo intravenoso para las mucopolisacaridosis I, II, IV y VI; este tratamiento no atraviesa la barrera hematoencefálica. El trasplante de progenitores hematopoyéticos está indicado en algunos casos de MPS VII y de MPS tipo I (en menores de 2,5 años y utilizando conjuntamente el tratamiento enzimático sustitutivo).
Defectos congénitos de glicosilación de proteínas (CDG)
La correcta glicosilación (adición de cadenas de azúcares, también denominados glicanos u oligosacáridos) a proteínas o lípidos es esencial para el desempeño adecuado de la función biológica de las glicoproteínas y de los glicolípidos. Las glicoproteínas son esenciales para funciones estructurales, de transporte, inmunológicas, hormonales, señal célula-célula y enzimáticas(12).
Han sido identificados cerca de 40 tipos de CDG, que son distribuidos en cuatro grandes grupos:
1. Defectos de la N-glicosilación.
2. Defectos de la O-glicosilación.
3. Defectos de la glicosilación de los esfingolípidos y de la fijación del glicosilfosfatidilinositol.
4. Defectos en la glicosilación múltiple y otras vías metabólicas.
La herencia es autosómica recesiva, excepto en el síndrome de las exóstosis múltiples cartilaginosas, de herencia autosómica dominante y los defectos de MAGT1 y ALG13, ligados al cromosoma X.
Las manifestaciones clínicas son multisistémicas y heterogéneas. La gran variabilidad de las manifestaciones clínicas y de su severidad, incluso dentro del mismo tipo de CDG y de la misma familia, debe ser considerada en el momento del consejo genético.
Se asocian a gran morbimortalidad, especialmente en la primera infancia.
La forma más frecuente, 80% de los casos de defecto de la N-glicosilación, es el CDG Ia, por defecto de la enzima fosfomanomutasa 2 (PMM2), debido a mutaciones en el gen PMM2 (cromosoma 16 p13). Presenta gran variabilidad en la expresión clínica. Inicialmente, durante los primeros meses de vida, domina el cuadro general, con fallo para medrar y dificultades para la alimentación. Posteriormente, predominan las manifestaciones neurológicas, como: movimientos lentos y erráticos de los ojos, movimientos lentos de la cabeza, estrabismo convergente, hipotonía axial, hiporreflexia, hipoplasia de cerebelo, temblor, desequilibrio y retraso psicomotor, más evidente desde los 4 meses de edad. Es frecuente la asociación de un fenotipo peculiar con: mejillas prominentes, orejas grandes y displásicas, distribución anómala de la grasa, mamilas invertidas, piel en cáscara de naranja y alteraciones esqueléticas. Entre las manifestaciones multisistémicas, destacan: derrame pericárdico, episodios stroke-like por aumento de la agregación plaquetaria, tubulopatía proximal, hipogonadismo, retinopatía pigmentaria, osteopenia y hepatopatía. La hepatopatía puede manifestarse con: hepatomegalia, elevación de transaminasas, hipoalbuminemia, coagulopatía, esteatosis, fibrosis y cirrosis. La mortalidad es elevada antes de los 2 años. Las alteraciones bioquímicas más frecuentes son: hipoalbuminemia, elevación de transaminasas, hipocolesterolemia e hipotrigliceridemia y disminución de antitrombina III, proteínas C y S, factor VIII y XI. Desde el punto de vista hormonal, aumento de FSH, LH, prolactina y TSH, con disminución de T4 libre.
El tipo CDG Ib, se debe a la deficiencia de fosfomanosa isomerasa (MPI) debida a mutaciones en el gen MPI del cromosoma 15. Cursa sin afectación neurológica y con predominio de afectación hepática e intestinal: vómitos recurrentes y diarrea, enteropatía exudativa pierde proteínas, hepatomegalia, disfunción hepática e incluso insuficiencia hepática. Hipoglucemia hiperinsulinémica. Dispone de tratamiento eficaz.
Las CDG plantean diagnóstico diferencial con las enfermedades mitocondriales. Ante la sospecha de CDG, se cuantifica la transferrina deficiente en carbohidratos (CDT) en suero o, más actualmente, se utiliza el EIE (enfoque isoeléctrico) de la transferrina o técnicas de electroforesis capilar. El diagnóstico se confirma mediante la determinación de actividad enzimática en leucocitos (en los defectos de PMM2 y de MPI) y en fibroblastos en el resto de los tipos. La identificación del defecto genético es importante para el consejo genético y el diagnóstico prenatal.
La mayoría de los CDG no dispone de tratamiento eficaz. El tratamiento con Manosa oral en CDG Ib, mejora la sintomatología, la coagulopatía, la enteropatía pierde proteínas y la hipoglucemia, pero se desconoce su efecto sobre la hepatopatía. En la CDG IIc, por defecto de transportador de la GDP-fucosa, está indicada la fucosa, que mejora las infecciones recurrentes, pero no las manifestaciones neurológicas.
En la PMM2-CDG (CDG Ia), el único tratamiento disponible es el sintomático; está indicado el soporte nutricional (con sonda nasogástrica o gastrostomía percutánea), el tratamiento antirreflujo gastroesofágico, el tratamiento hormonal del hipogonadismo y el empleo de bifosfonatos en casos de fracturas recurrentes. El empleo de ácido acetilsalicílico para la prevención de fenómenos stroke-like, debe ser cuidadosamente evaluado, ponderando el riesgo trombótico frente al hemorrágico, ya que estos pacientes tienen riesgo aumentado de ambos fenómenos.
Enfermedades peroxisomales
Los peroxisomas son organelas intracelulares que contienen diferentes enzimas catabólicos y anabólicos(13). Los trastornos peroxisomales son de herencia autosómica recesiva, excepto la adrenoleucodistrofia ligada al X. Se clasifican en dos grandes grupos:
1. Grupo de desórdenes de la biogénesis peroxisomal, la organela fracasa en su formación, existe una alteración severa del número o morfología de los peroxisomas y una deficiencia de múltiples funciones. Se debe a mutaciones en los genes PEX y alteración de sus productos, peroxinas. Comprenden la condrodisplasia rizomiélica punctata tipo I y el “espectro Zellweger”, que incluye: el síndrome de Zellweger, la adrenoleucodistrofia neonatal y la enfermedad de Refsum infantil.
2. Grupo de desórdenes por defectos enzimáticos aislados (déficit de un único enzima peroxisomal con la estructura peroxisomal intacta o poco modificada). Incluye la adrenoleucodistrofia ligada al X, que es la enfermedad peroxisomal más frecuente, con afectación neurológica y suprarrenal.
El Síndrome de Zellweger debuta en el período neonatal con: rasgos dismórficos, anomalías neurológicas severas, retinopatía y trastorno hepático, (hepatomegalia con progresión a cirrosis) que queda enmascarado por el predominio de las alteraciones neurológicas; pueden presentar quistes renales y calcificaciones puntiformes epifisarias. Fallecen en el primer año de vida. Los datos bioquímicos que orientan al diagnóstico son: la elevación de ácidos grasos de cadena muy larga, de ácido fitánico y pristánico y de ácido pipecólico, con disminución de plasmalógenos. El diagnóstico se confirma por estudio enzimático en fibroblastos. El estudio de microscopía electrónica de los tejidos muestra ausencia de peroxisomas. Es posible el diagnóstico prenatal. El tratamiento es de soporte.
Tumores hepáticos
Los tumores hepáticos más frecuentes son: hepatoblastoma, hepatocarcinoma y hemangiomas, representan dos tercios de los tumores hepáticos pediátricos.
Hepatoblastoma
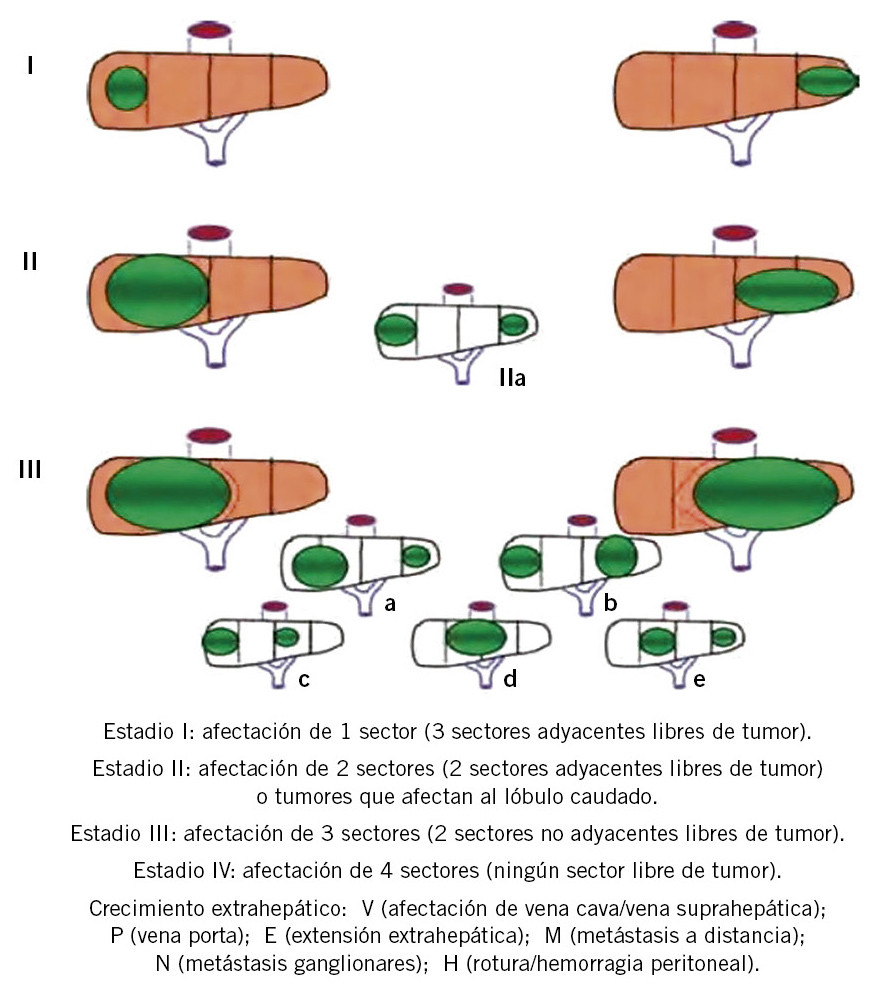
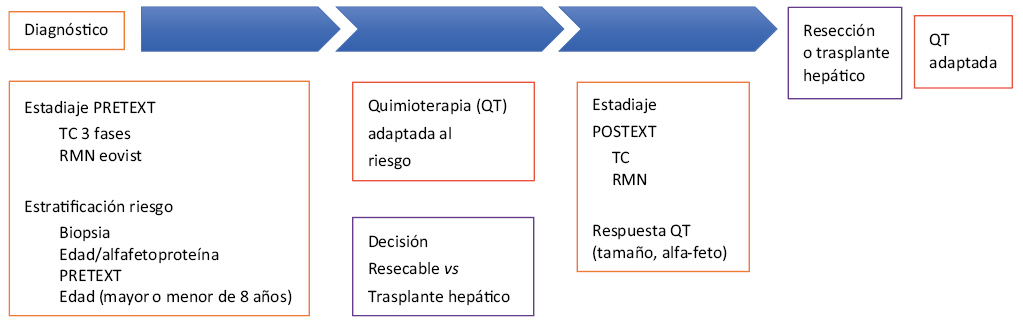
Es el tumor hepático más frecuente en la infancia, supone el 80% de todos los tumores malignos hepáticos(14,15). Afecta preferentemente a varones, entre los 6 meses y 3 años, y es rara la presentación neonatal. Se ha descrito asociación con el síndrome de Beckwith-Wiedemann (cromosoma 11), la poliposis adenomatosa familiar y el síndrome de Gardner (cromosoma 5). La prematuridad y el bajo peso al nacimiento (<1.000 g) son considerados factores de riesgo. La forma habitual de presentación es la detección de hepatomegalia o masa abdominal asintomática; es muy poco frecuente que exista alteración de la función hepática, aunque un 5% presentan ictericia. En un 20%, se objetivan metástasis al diagnóstico, la mayoría pulmonares. En el 90% de los casos, existe elevación de alfa-feto proteína; su determinación es de gran ayuda en el diagnóstico, en la monitorización de la respuesta al tratamiento y en la detección precoz de la recurrencia tumoral. La ecografía permite el diagnóstico en la mayoría de los casos, siendo necesario completar el estudio con TAC tóraco-abdominal para valorar la extensión. Está indicada la biopsia del tumor; histológicamente, se diferencian los tipos, epitelial, mixto, anaplásico y macrotrabecular. En el tipo epitelial, se distinguen varios patrones, siendo el fetal el de evolución más favorable. La indiferenciación histológica se asocia con pronóstico desfavorable. Actualmente, el sistema de estadiaje más empleado es el PRETEXT (PRE-treatment EXT-ension), basándose en las pruebas de imagen realizadas en el momento del diagnóstico. Distingue cuatro estadios tumorales (PRETEXT I-IV), dependiendo del número de segmentos hepáticos afectados, y expresa mediante una letra criterios adicionales(12), como: afectación del lóbulo caudado (C), extensión extrahepática abdominal (E), focalidad del tumor (F), ruptura tumoral o hemorragia intraperitoneal (H), metástasis a distancia (M), afectación de ganglios linfáticos (N) y afectación vascular de vena cava o suprahepática (V) y de vena porta (P). Son considerados pacientes de alto riesgo(16) los que presentan alguno de los siguientes datos: alfafetoproteína < 100 ng/ml, PRETEXT IV o cualquier grado de PRETEXT si existe: extensión intraabdominal extrahepática, nódulos peritoneales, metástasis, rotura tumoral o hemorragia intraperitoneal o afectación vascular (porta principal o sus ramas, afectación de 3 venas suprahepáticas y/o cava inferior). El tratamiento consiste en la administración de quimioterapia seguida de la resección quirúrgica del tumor y quimioterapia postquirúrgica. El trasplante hepático(16) está indicado en los casos considerados irresecables tras administración de quimioterapia y que no tienen afectación extrahepática. La tasa de curación aproximada actualmente es del 75%.
Hepatocarcinoma
Es un tumor de alta malignidad. Solo un 33% de los niños occidentales con hepatocarcinoma presentan una enfermedad de base que se asocie al desarrollo de tumor (atresia biliar, colestasis intrahepática familiar, defecto de alfa-1-antitripsina, síndrome de Alagille, tirosinemia, glucogenosis o hepatitis B y C). Afecta a niños mayores de 10 años, con predominio en varones(17,18). La forma de presentación habitual es la palpación de masa abdominal. Si asienta sobre una enfermedad que cursa con cirrosis, los síntomas predominantes serán los de la hepatopatía crónica. El diagnóstico se realiza de forma similar al del hepatoblastoma, con la determinación de alfa-fetoproteína (elevada solo en 50-70% de los pacientes), ecografía, TAC y biopsia. Existen dos patrones histológicos, el anaplásico o epitelial y el fibrolamelar. El 25% de los niños con hepatocarcinoma tienen metástasis en el momento del diagnóstico, la mayoría en ganglios abdominales y mediastínicos y en los pulmones. El estadiaje es realizado mediante el sistema PRETEXT. El tratamiento se basa en la quimioterapia antes y después de la resección quirúrgica; en estudio, la talidomida y el sorafenib. El trasplante está indicado en los irresecables sin extensión extrahepática y en los hepatocarcinomas que asientan sobre enfermedad hepática con cirrosis. El pronóstico es malo, con supervivencia del 20-30% a los 5 años del diagnóstico.
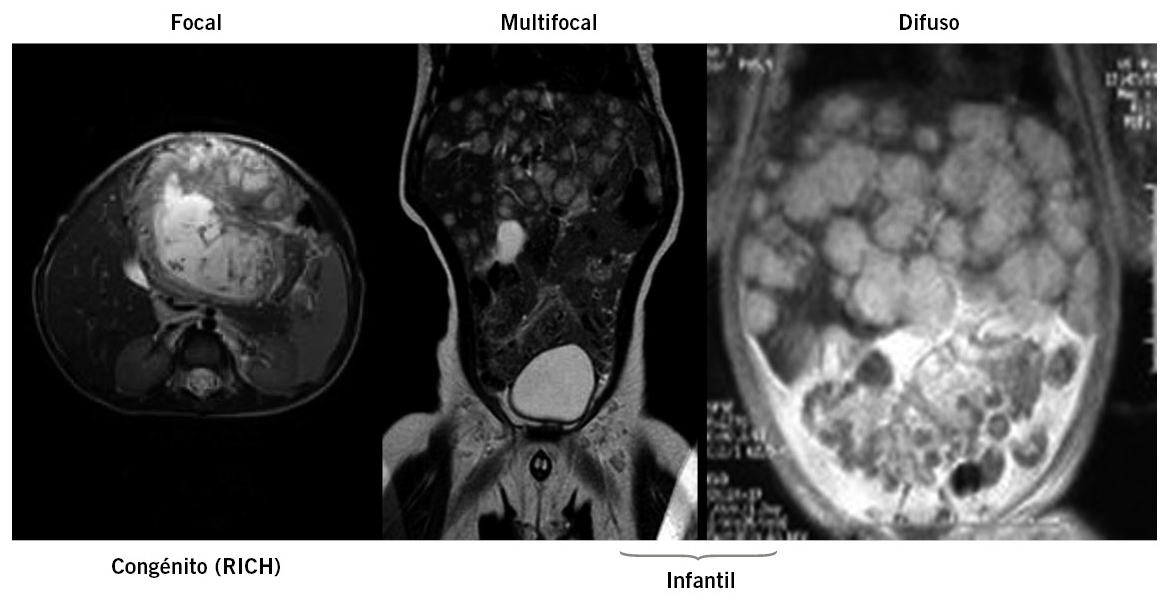
Hemangiomas hepáticos infantiles
Tumores hepáticos histológicamente benignos, pero que pueden asociar morbimortalidad significativa.
Tres subtipos(17,18): hemangioma focal o hepático solitario, multifocal (el más frecuente) y difuso. El hemangioma hepático solitario es un RICH (rapidly involuting congenital hemangioma), con involución rápida antes del año; la detección es prenatal o al nacimiento. Suelen ser asintomáticos, pero pueden existir fístulas intraparenquimatosas y desarrollar insuficiencia cardíaca. Pueden provocar trombocitopenia y anemia. No expresan el marcador inmunohistoquímico GLUT- 1.
El hemangioma hepático multifocal presenta una fase de crecimiento rápida tras el nacimiento (duración 1 año, prolifera sobre todo en los primeros 4 meses), seguida de una fase involutiva; es GLUT-1 positivo. A menudo son asintomáticos, pero puede existir insuficiencia cardíaca e hipotiroidismo. Puede aparecer en el contexto de una hemangiomatosis neonatal difusa (múltiples hemangiomas eruptivos en piel y mucosas, pueden asociar hemangiomas hepáticos, pulmonares, gastrointestinales o intracraneales).
El hemangioma hepático difuso tiene características similares al multifocal, es GLUT-1 positivo. Se asocia a un curso más agresivo con hepatomegalia masiva (afecta a la totalidad del parénquima hepático) y puede provocar un síndrome compartimental abdominal a los 3-4 meses con compromiso respiratorio, compromiso del retorno venoso y compresión de la vena renal. Es posible el desarrollo de insuficiencia hepática. Otra complicación es el hipotiroidismo debido a sobreproducción de iodotironina deiodinasa tipo 3, que degrada la hormona tiroidea.
El diagnóstico se basa en la clínica y en las pruebas de imagen (ecografía doppler, angioRMN, angioTAC); no está indicada la biopsia por el riesgo de sangrado, salvo en caso de patrón radiológico anómalo. Es necesario realizar ecografías seriadas a lo largo del primer año.
Tratamiento. El hemangioma focal no responde a propanolol ni a corticoides. La mayoría desaparece en el primer año, por lo que se recomienda tratamiento conservador; pueden requerir embolización o hepatectomía parcial en caso de shunts con insuficiencia cardíaca. En los hemangiomas multifocales y en los difusos, está indicado el tratamiento de las complicaciones asociadas, como insuficiencia cardíaca e hipotiroidismo. El manejo ha cambiado en los últimos años tras describirse la eficacia y buena tolerancia al propanolol en ambos tipos de hemangiomas. Los hemangiomas difusos tienen un comportamiento más agresivo, es recomendable el ingreso en Cuidados Intensivos. En algunos casos refractarios difusos, puede estar indicado el trasplante hepático o la embolización. Anteriormente, se han utilizado otros tratamientos como el interferón, quimioterapia y corticoides.
Síndrome de Budd-Chiari(19)
El síndrome de Budd-Chiari es debido a una obstrucción al flujo venoso hepático de origen no cardiogénico, que condiciona la aparición de ascitis y hepatomegalia.
En niños, a diferencia de los adultos, puede cursar solo con hepatomegalia. La oclusión de las venas suprahepáticas o de la cava suprahepática se puede producir en el contexto de diversas situaciones que predisponen a la trombosis, como: trastornos linfoproliferativos, invasión tumoral, traumatismo abdominal, defectos de proteínas C, S y antitrombina III, hiperhomocisteinemia, mutación del factor V Leyden, policitemia vera, enfermedad de Behcet, embarazo, anticonceptivos orales, colagenosis, síndrome antifosfolípido, hemoglobinuria paroxística nocturna y anemia de células falciformes. Otro mecanismo que causa obstrucción al flujo venoso es la presencia de lesiones congénitas, como la estenosis o hipoplasia de venas suprahepáticas o la obstrucción membranosa de la cava suprahepática (puede asociarse a infecciones o a estados de hipercoagulabilidad). La insuficiencia cardíaca derecha y la pericarditis constrictiva pueden desencadenar un cuadro parecido al síndrome de Budd-Chiari. La clínica del síndrome de Budd-Chiari depende de la rapidez de instauración de la oclusión y de su extensión. La instauración brusca desencadena una forma de presentación fulminante, con insuficiencia hepática aguda, mientras que la instauración lenta desencadena el desarrollo de una hepatopatía crónica. En la forma aguda, la presentación clínica consiste en: dolor abdominal, vómitos, hepatomegalia y ascitis con evolución a fallo hepático. En la forma crónica, el dolor abdominal, hepatomegalia y ascitis se desarrollan en 1-6 meses, puede aparecer esplenomegalia. Desde el punto de vista analítico, es característica una leve elevación de transaminasas y de bilirrubina, con hipoalbuminemia marcada; en las formas agudas, existe coagulopatía, con elevación importante de transaminasas, y en las formas crónicas, hiperesplenismo. La ecografía doppler puede revelar flujo monofásico de las suprahepáticas o alteraciones en la cava, pero en la mayoría de las ocasiones se requieren pruebas de imagen más finas, como el angioTAC o angioRMN. La cavografía permite detectar el nivel y extensión de la obstrucción y, en ocasiones, su tratamiento mediante angioplastia. Está indicado realizar ecocardiograma para descartar problema cardíaco. La biopsia hepática, solo posible en situaciones sin insuficiencia, revela intensa congestión centrolobulillar con necrosis y dilatación de los sinusoides, con grados variables de fibrosis. El tratamiento consiste en el manejo de la ascitis; es necesario descartar obstrucción o estenosis susceptible de corrección mediante angioplastia o cirugía. En los casos que se presentan ya con cirrosis o con fallo hepático agudo, el tratamiento de elección es el trasplante hepático, siempre que no existan contraindicaciones.
Función del pediatra de atención primaria
• La hepatomegalia es un signo físico detectado con relativa frecuencia en la consulta del pediatra que no debe ser menospreciado.
• El proceso diagnóstico ante una hepatomegalia incluye:
- Una minuciosa anamnesis.
- Exploración física completa (no solo abdominal).
- Percentiles de peso y talla.
- Pruebas complementarias de primer nivel (analítica completa con coagulación y función hepática; ecografía doppler abdominal).
• Esta orientación inicial nos permite discernir, en la mayoría de las ocasiones, si nos enfrentamos a un proceso agudo o crónico, si el paciente presenta una hepatopatía o una enfermedad sistémica que cursa con hepatomegalia y si es posible el manejo ambulatorio o precisa asistencia hospitalaria (urgente o programada).
• Los pacientes que presenten signos clínicos de gravedad o compatibles con proceso tumoral y los recién nacidos con hepatomegalia y colestasis deben ser remitidos a un centro hospitalario con el fin de agilizar el diagnóstico y tratamiento. Si hay sospecha clínica y/o analítica de fallo hepático, deben ser remitidos de manera inmediata a un hospital con equipo de trasplante hepático.
• En casos que no sugieran gravedad, se puede iniciar el estudio en Atención Primaria, realizando los estudios de primer nivel lo más rápidamente posible y considerando posteriormente su derivación a consultas hospitalarias.
Bibliografía
Los asteriscos reflejan el interés del artículo a juicio del autor.
1.*** Wolf AD, Lavine JE. Hepatomegaly in Neonates and Children. Pediatrics in Review. 2000; 21(9): 303-10. Interesante revisión con enfoque práctico del abordaje de la hepatomegalia en el paciente pediátrico.
2.*** Frauca Remacha E, Muñoz Bartolo G. Colestasis en el lactante. En: Protocolos Diagnósticos-Terapéuticos de Gastroenterología, Hepatología y Nutrición Pediátrica. ISBN 978-84-8473-869-5. Madrid: Ergon. 2010; p. 177. Revisión de las causas más frecuentes de colestasis en el lactante, diagnóstico y tratamiento.
3.*** Chakrapani A, Green A. Metabolic liver disease in the infant and older children. Diseases of the liver and biliary system in children. En: Kelly DA, ed. Blackwell Publishing Ltd; 2004. Interesante capítulo resumiendo los principales trastornos metabólicos en la infancia que cursan con hepatomegalia. Tablas resumen muy prácticas.
4.*** Kido J, Nakamura K, Matsumoto S et al. Current status of hepatic glycogen storage disease in Japan: clinical manifestations, treatments and long-term outcomes. Journal of Human Genetics. 2013; 58: 285-92. Revisión completa de 127 glucogenosis, detallando las manifestaciones clínicas, tratamiento y pronóstico a largo plazo.
5.*** Kishnani P, Austin S, Abdenur J et al. Diagnosis and management of glycogen storage disease type I: a practice guideline of the American College of Medical Genetics and Genomics. Genet in Medicine. 2014; 10: 1038. Revisión actual sobre el manejo de las glucogenosis.
6. Boers S, Visser G, Smit P and Fuchs S. Liver transplantation in glycogen storage disease type 1. Orphanet Journal of Rare Diseases. 2014; 9: 47.
7.*** Hierro L. Indicadores de pronóstico y establecimiento de la indicación de trasplante. En: Paloma Jara, editor. Trasplante hepático en niños. Madrid: Ergon. 2006; p. 91-116. Criterios de indicación de trasplante en las diferentes hepatopatías de la infancia y revisión de la evolución y el pronóstico de las mismas.
8.*** Díaz Fernández MC. Indicaciones y contraindicaciones de trasplante. En: Paloma Jara, editor. Enfermedad hepática en el niño. ISBN-13:978-84-938973.
9. Elstein D, Zimran A. Review of the safety and efficacy of imiglucerase treatment of Gaucher disease. Biologics: Targets & Therapy. 2009; 3: 407-17. Interesante revisión sobre el tratamiento de la enfermedad de Gaucher con imiglucerasa, abordando los objetivos terapéuticos y la eficacia, así como nuevas opciones terapéuticas.
10. Pineda Marfá M, Coll Rosell MJ. Enfermedad de Niemann-Pick tipos A, B y C. En: Sanjurjo P, Baldellou A, eds. Diagnóstico y tratamiento de las enfermedades metabólicas hereditarias. 4ª edición. Madrid: Ergon. 2014; p. 961-72. Capítulo actualizado sobre los tres tipos de la enfermedad de Niemann-Pick.
11. ***Vanier MT. Niemann-Pick disease type C. Orphanet Journal of Rare Diseases. 2010; 5: 16. Revisión actualizada de los aspectos clínicos, diagnósticos y terapéuticos de la enfermedad de Niemann-Pick C.
12. ***Vilarinho L, Azevedo L, Leao Teles E. Defectos congénitos de la glicosilación. En: Sanjurjo P, Baldellou A, eds. Diagnóstico y tratamiento de las enfermedades metabólicas hereditarias. 4ª edición. Madrid: Ergon. 2014; p. 1039-55. Actualización detallada de los defectos congénitos de la glicosilación.
13. ***Spécola N. Diagnóstico de las enfermedades peroxisomales. En: Sanjurjo P, Baldellou A, eds. Diagnóstico y tratamiento de las enfermedades metabólicas hereditarias. 4ª edición. Madrid: Ergon. 2014; p. 1021-29. Capítulo actualizado y conciso sobre las enfermedades peroxisomales.
14. ***Frauca E. Tumores hepáticos. En: Paloma Jara, editor. Enfermedad hepática en el niño. Madrid: Ergon. ISBN-13:978-84-938973-3-8. P. 515-34. Revisión y actualización del estadiaje y tratamiento de los principales tumores hepáticos en la infancia.
15. ***Otte JB. Progress in the surgical treatment of malignant liver tumors in children. Cancer Treatment Reviews. 2010; 36: 360-71. Artículo reciente en el que se revisa el estadiaje y las opciones terapéuticas del hepatoblastoma y el hepatocarcinoma.
16. ***Barrena S, Hernández F, Miguel M et al. High-Risk Hepatoblastoma: Results in a Pediatric Liver Transplantation Center. Eur J Pediatr Surg. 2010 Oct 11. Revisión de los resultados en el tratamiento de hepatoblastomas de alto riesgo en el Hospital Infantil La Paz.
17. ***López Almaraz R, López Gutiérrez JC, Belendez Bieler C, Herrero Hernández A, Mateos González ME y Ramírez Villar G. Tumores vasculares en la infancia. An Pediatr (Barc). 2009. doi:10.1016/j.anpedi.2009.10.007. Interesante artículo del Grupo de Tumores poco frecuentes de la Sociedad Española de Hematología y Oncología Pediátricas (SEHOP)-Subcomité Tumores Vasculares, en el que se clarifica la nomenclatura y la clasificación de las lesiones vasculares en la infancia, abordando su diagnóstico y tratamiento.
18. ***Christison-Lagay ER, Burrows PE, Alomari A et al. Hepatic hemangiomas: subtype classification and development of a clinical practice algorithm and registry. Journal of Pediatric Surgery. 2007; 42: 62-8. Clasificación de los hemangiomas hepáticos, con algoritmo diagnóstico y terapéutico.
19. Karrel MK and Bucuvalas JC. Systemic Disease and the liver. En: Suchy FJ, Sokol RJ, Balistreri WF ed. Liver Disease in Children. Third edition. Philadelphia: Lippincot Williams & Wilkins. 2007; p. 897-927. Explicación clara y breve del S. Budd-Chiari en la infancia.
| Caso clínico |
|
Motivo de consulta
Varón de 2 meses y medio. Ingresa, trasladado desde otro centro hospitalario, por colestasis y hepatoesplenomegalia.
Antecedentes familiares
Padres no consanguíneos. Sin interés.
Antecedentes personales
Embarazo controlado, curso normal. Parto eutócico. EG: 37 + 4 semanas. PRN 2,550 kg, talla: 48 cm. No reanimación. Diuresis y meconio en primeras 24 horas. Ictericia neonatal, precisó fototerapia durante 5 días (bilirrubina máxima 17 mg/dl). Pruebas metabólicas no recibidas (cambio de domicilio).
Lactancia materna durante 1 mes, posteriormente artificial exclusiva con fórmula de inicio. No introducción de sacarosa. Buena tolerancia, aunque siempre con dificultad para realizar las tomas. Orina y deposiciones (4-5/día) referidas como normales. No procesos intercurrentes. Sonrisa social. No síntomas de hipoglucemia. Duerme bien. Distensión abdominal desde el período neonatal.
Enfermedad actual
Los padres refieren aumento de la ictericia y la distensión abdominal en los últimos días, irritabilidad y rechazo de las tomas.
Exploración física
Peso 4,120 kg (< P 3), talla: 56 cm (P 3-10).
REG. Malnutrición importante, masas musculares escasas, ausencia de panículo adiposo. Ictericia leve de piel y mucosas. No dificultad respiratoria. Rinorrea clara. No craneotabes. ACP: rítmico, no soplos. Pulsos palpables y simétricos. Buena ventilación bilateral, ruidos transmitidos de vías altas. ABD: gran distensión abdominal, circulación colateral abdominal prominente. Hepatoesplenomegalia de consistencia dura, hasta fosas ilíacas. Ascitis. Hidrocele bilateral, no complicado. NRL: fontanela anterior normotensa, sonrisa social. Deposiciones con color.
Pruebas complementarias de primer nivel
Hemograma: Hb: 13,2 g/dl, Hcto: 40,9%, Leu: 15.310 (N: 34, L: 56, M: 4, Eo: 3, cayados: 1, eritroblastos: 2%). Plaquetas: 219.000, VSG: 9.
Coagulación: APP: 93%, tiempo de cefalina: 35,6“ (1,19), fibrinógeno: 286.
Bioquímica: AST: 378, UI/l, ALT: 74, UI/l, GGT: 326 UI/l, fosfatasa alcalina: 186 UI/l, bilirrubina: 10 mg/dl, bilirrubina directa: 5 mg/dl, colesterol: 190 mg/dl, triglicéridos: 337 mg/dl, colinesterasa: 3.935 UI/l, urato: 2,5 mg/dl, creatinina: 0,4 mg/dl, Glu: 63 mg/dl, proteínas totales: 6,5 g/dl, albúmina: 4,1 g/dl, LDH: 563 UI/l, gasometría: normal. Láctico normal.
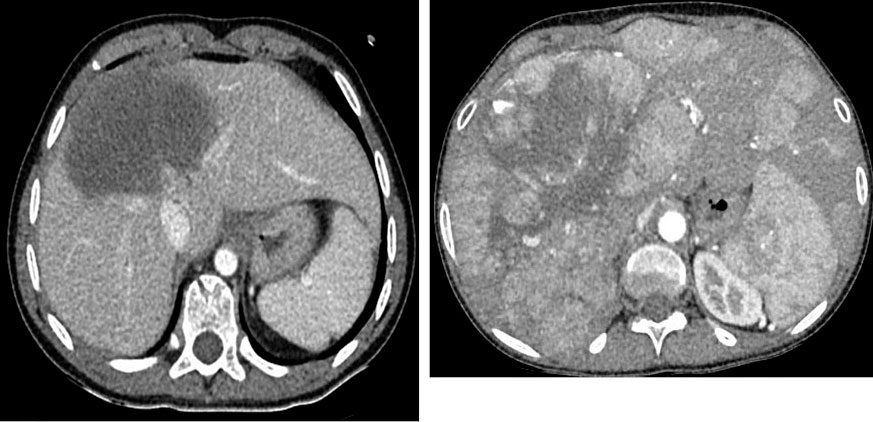
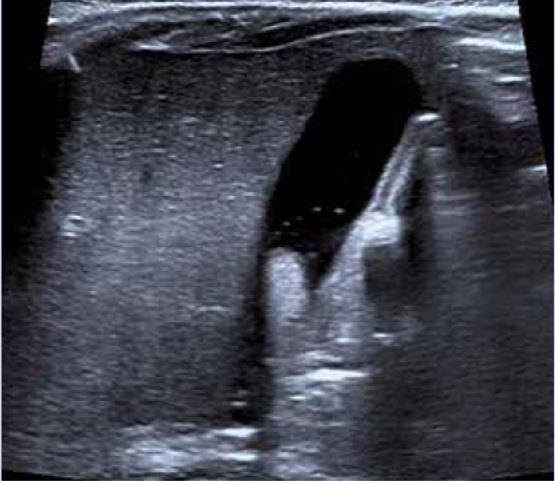
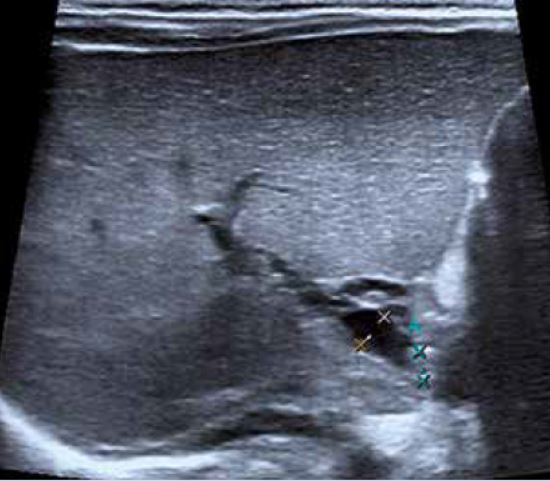
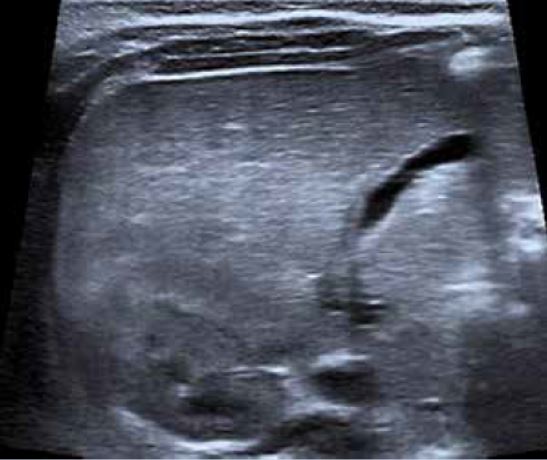
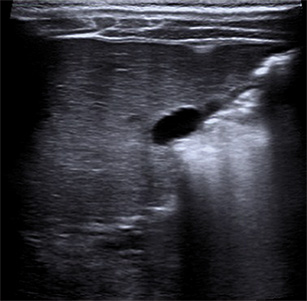
Ecografía abdominal: hígado aumentado de tamaño, parénquima homogéneo. Vesícula normal, vía biliar intra y extrahepática no dilatadas. Esplenomegalia homogénea de 9,5 cm. Vena porta permeable con flujo hepatópeto de 20 cm/s. Arteria hepática permeable con índice resistencia 0,78. Venas suprahepáticas permeables con patrón espectral trifásico. Riñones de tamaño normal, con aumento de la ecogenicidad parenquimatosa y mala diferenciación córtico-medular; imágenes sugestivas de quistes medulares y en la unión córtico-medular, milimétricos y puntiformes. No dilatación de la vía excretora. Pequeña cantidad de líquido libre perihepático y periesplénico.
Ecocardiograma: normal.
Evolución
Al ingreso, presenta malnutrición importante, hepatoesplenomegalia masiva, ascitis, las deposiciones no son acólicas. El estudio de primer nivel, muestra colestasis con GGT elevada, aumento de transaminasas, sin signos de insuficiencia hepática y sin datos de hiperesplenismo. En la ecografía, se observa flujo trifásico en las suprahepáticas y flujo portal normal. Se descarta origen cardiológico de la hepatoesplenomegalia.
La sospecha diagnóstica inicial es de enfermedad de depósito, dada la gran hepatoesplenomegalia con ascitis y la ausencia de signos ecográficos de tumor hepático, de hipertensión portal o de síndrome de Budd-Chiari (confirmada normalidad de suprahepáticas y cava en TAC realizado posteriormente). Es necesario descartar procesos tumorales extrahepáticos, infecciosos u otras enfermedades metabólicas y, con este objetivo, se realizan las siguientes pruebas complementarias:
- Rx abdomen: sin calcificaciones suprarrenales.
- Rx esqueleto: distensión abdominal con centralización de las asas (ascitis). Edad ósea corresponde a un paciente recién nacido. Osteoporosis. Ligero acopamiento de la metáfisis distal de cúbito y radio.
- Alfa-fetoproteína: 176.689 ng/ml.
- Catecolaminas en orina: normales.
- Lactato, piruvato y cociente lactato/piruvato: normal. No se detecta beta hidroxibutirato.
- Galactosa-1-P-uridiltransferasa: actividad normal.
- Estudio de ácidos biliares en orina normal. CDT normal. Mucopolisacáridos en orina negativos. No excreción de succinilacetona. Aminoácidos y ácidos orgánicos en sangre y orina: normales.
- Serología virus: anti-VHC (–), HBsAg (–), anti-core (–), anti-HBs (+), VIH (–).
- Serología infección connatal: IgG sífilis (–), IgM toxoplasma (–), IgM rubeola (–) IgM CMV (+).
- PCR EBarr: negativa. Antigenemia CMV: 66/200.000 PMN.
- Estudio oftalmológico: posible mancha rojo cereza.
- Ecografía cerebral: sin calcificaciones. Normal.
A los 4 días del ingreso, antes de obtener el resultado de algunas de las pruebas anteriores, presenta sepsis de origen urinario (E. Coli), requiriendo ingreso en Cuidados Intensivos Pediátricos, con soporte inotrópico y ventilación mecánica. Evoluciona con importante ascitis que condiciona compromiso renal y respiratorio, con incremento de la presión intraabdominal, precisando paracentesis evacuadora y tratamiento diurético agresivo.
Simultáneamente, se conoce resultado de antigenemia CMV positiva (66/200.000 PMN), por lo que se asocia al tratamiento antibiótico, tratamiento antiviral con Ganciclovir iv. Posteriormente, presenta neumonía por St aureus, Enterobacter cloacae y Stenotrophomonas maltophilia. En el contexto de infección respiratoria y dificultad para la extubación, se realiza TAC torácico, que muestra afectación intersticial radiológicamente inespecífica con presencia de áreas de atenuación en vidrio deslustrado. Es realizado estudio inmunológico que descarta inmunodeficiencia.
Es continuado el estudio etiológico, realizándose otras pruebas complementarias orientadas al diagnóstico de enfermedades de depósito: biopsia de médula ósea, biopsia de piel para determinación de actividades enzimáticas en cultivo de fibroblastos y análisis de actividad enzimática en leucocitos. El proceso séptico retrasa la realización de biopsia hepática.
Médula ósea (11 días postingreso): celularidad aumentada y aspecto reactivo. Muy escasos histiocitos de aspecto espumoso con escasas imágenes de eritrofagocitosis, sin que se identifiquen microorganismos en su interior. PCR Leishmania en médula ósea: negativa.
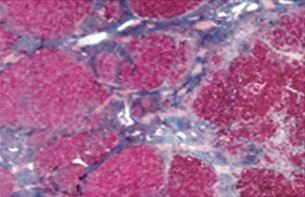
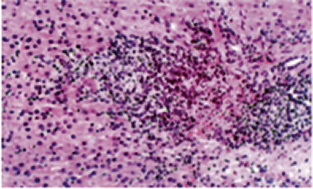
Biopsia hepática (26 días postingreso): arquitectura general alterada por el depósito en células de Küppfer, macrófagos portales y hepatocitos salpicados, de un material espumoso, con vacuolas ópticamente vacías y un fino reticulado débilmente eosinófilo. Con técnica PAS, este reticulado es positivo, aunque mucho menos que el contenido glucogénico normal hepatocitario, y se mantiene tras el tratamiento con diastasa. No se reconocen depósitos de colesterol (técnica de Schultz); con técnicas de Oil-Red O y Sudán negro, se identifican depósitos focales globulares en hepatocitos y células del sistema retículo endotelial. No hay depósitos de hierro, signos de colestasis ni otras alteraciones. Con técnica de Masson, se observa una ligera expansión fibrosa estrellada de los espacios porta y de algunos sinusoides, no puenteante. Hay nidos de hematopoyesis. No se observan inclusiones virales, tampoco con inmunohistoquímica para CMV. Microscopía electrónica: Presencia de abundantes estructuras membranosas laminares en ocasiones concéntricas, que recuerdan a las figuras de mielina y que se identifican en hepatocitos y células retículoendoteliales. Diagnóstico: Hígado con alteraciones sugestivas de enfermedad de depósito lisosomal.
Actividad enzimática de Esfingomielinasa y Beta-glucocerebrosidasa en leucocitos: normal.
Cultivo de fibroblastos (11 días postingreso): actividad de lipasa ácida normal. Test de filipina: acúmulo de vesículas perinucleares fluorescentes (colesterol libre). Este resultado indica que el paciente está afecto de enfermedad de Niemann-Pick C en su forma bioquímica clásica. El estudio genético confirma el diagnóstico (mutación en el gen NPC1).
El diagnóstico de nuestro paciente es, por tanto, enfermedad de Niemann-Pick tipo C, de inicio perinatal, sin afectación neurológica en el momento del diagnóstico y con afectación visceral grave.
Evolución: el paciente requiere ventilación mecánica durante 47 días y posteriormente oxigenoterapia de alto flujo, permanece ingresado durante 2 meses en Intensivos Pediátricos. La afectación respiratoria es debida en parte al patrón intersticial radiológico y, sobre todo, al componente restrictivo derivado de la gran visceromegalia y de la ascitis. La infección por CMV puede haber contribuido a la elevación inicial de transaminasas y a la afectación respiratoria. Desde el punto de vista hepático, no desarrolla signos de insuficiencia hepática ni signos de hiperesplenismo. Evolución favorable de la colestasis y la hipertransaminasemia, con normalización de bilirrubina a los 6 meses de edad, manteniendo elevación de GGT (500 UI/l) y discreta elevación de AST (< 150 UI/l). Mala evolución del paciente condicionada por la hepatoesplenomegalia y la ascitis con compromiso respiratorio secundario, requiriendo oxigenoterapia y, ocasionalmente, oxigenoterapia de alto flujo. Fallece a los 10 meses por cuadro de insuficiencia respiratoria.
El paciente recibe tratamiento de soporte con vitaminas liposolubles, ácido ursodeoxicólico y fenobarbital (hasta normalización de bilirrubina). Antibioterapia según necesidades, paracentesis evacuadoras de repetición y tratamiento diurético (furosemida, espironolactona). Oxigenoterapia y oxigenoterapia de alto flujo. Solicitado tratamiento con miglustat oral.
|
|
|