 |
| Temas de FC |
A. de la Vega, E. Frauca Remacha
Servicio de Hepatología y Trasplante Hepático.
Hospital Universitario Infantil La Paz. Madrid
| Resumen
Se revisa una de las formas habituales de presentación de la enfermedad hepática en Pediatría, con especial atención al diagnóstico diferencial según la edad del niño. Se describen los aspectos: clínicos, diagnósticos y terapéuticos de las etiologías más frecuentemente implicadas en la ictericia colestática en Pediatría. El espectro de posibles enfermedades responsables de su aparición es muy variado, y muy diferente según la edad del paciente. La gravedad de una colestasis la determinan la severidad de la lesión hepática y de sus consecuencias (insuficiencia o fallo hepático, hipertensión portal, encefalopatía, etc.) |
| Abstract
Review of a usual form of clinical presentation of hepatic disease in children, with special attention in differential diagnosis based upon children ́s age. The clinical, diagnostic, and therapeutic aspects of more frequently implied aetiologies in cholestatic jaundice in children are reviewed. There is a wide spectrum of diseases that can cause cholestasis, and this variety differs according the child ́s age. Conjugated jaundice importance depends on liver histological damage severity and its consequences (liver failure or insufficiency, portal hypertension, encephalopathy, etc.) |
Palabras clave: Ictericia; Colestasis
Key words: Jaundice; Cholestasis
Pediatr Integral 2015; XIX (3): 168-179
Síndrome colestático. Actitud diagnóstico-terapéutica
Introducción
La colestasis es una de las formas habituales de presentación de la enfermedad hepática en niños. No tiene ninguna especificidad desde el punto de vista diagnóstico.
La ictericia colestática, como único hallazgo inicial, es una de las formas habituales de presentación de las hepatopatías infantiles e, incluso, hay que tenerla en cuenta como primera manifestación de enfermedades no primariamente hepáticas. No tiene en si misma ninguna especificidad diagnóstica.
Es muy importante tener en cuenta que, el espectro de causas de enfermedad hepática y su habitual forma de debut es muy diferente según sea la edad del niño (Fig. 1). En el recién nacido o lactante, las causas de hepatopatía predominantes son: la inflamación idiopática (atresia biliar) y los trastornos genéticos (colestasis intrahepática, déficit de alfa-1-AT, metabolopatías, etc.); y la presentación habitual la de un cuadro de ictericia colestática que, en un porcentaje pequeño de casos, asocia una situación de fallo hepático. En el lactante (mayor de 6 meses) o niño mayor, las posibles causas de hepatopatía son más variadas (congénitas o adquiridas) y se detectan frecuentemente de forma casual o ante síntomas inespecíficos (astenia, anorexia, dolor abdominal, etc.). Es en este grupo de niños donde, con frecuencia, se detecta una elevación de transaminasas y, con menos frecuencia, una ictericia colestática, lo que constituye el punto de partida para el diagnóstico de su enfermedad hepática.
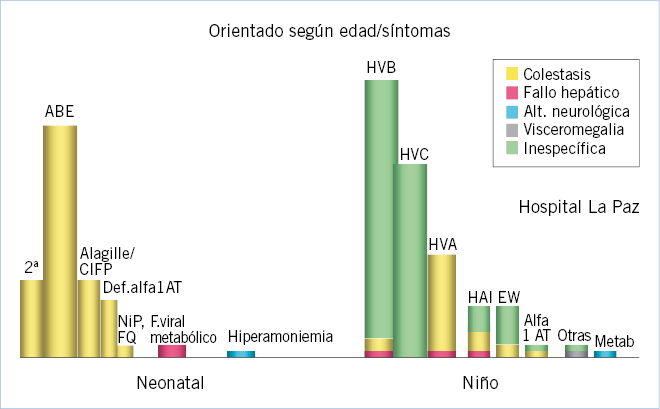
Figura 1. Enfermedades hepáticas en la infancia.
Por otra parte, las enfermedades hepáticas en la infancia pueden presentarse de formas diferentes (p. ej., la enfermedad de Wilson puede debutar como un fallo hepático agudo con ictericia o diagnosticarse en el estudio de un paciente tras el hallazgo de transaminasas elevadas) o bien debutar con una constante presentación clínica (p. ej., la atresia biliar como ictericia colestática en las primeras semanas de vida).
A lo largo de este tema, se irán describiendo las distintas entidades que pueden debutar como una ictericia colestática, pero teniendo en cuenta que, en muchas ocasiones, pudiera ser otra la forma de presentación (aumento de transaminasas, fallo hepático, etc.).
Concepto y recuerdo fisiopatológico
El término colestasis define aquella situación en la que existe una alteración del flujo biliar, con la consiguiente retención y paso a sangre de componentes de la bilis (bilirrubina directa, sales biliares, colesterol, etc.), y que condiciona un cuadro clínico característico, con: ictericia (tinte amarillento de piel y escleras), aparición de bilirrubina en orina (coluria), decoloración parcial o completa de las deposiciones (hipo o acolia) y prurito, y bioquímico, con: aumento de bilirrubina directa, GGT, fosfatasa alcalina y colesterol.
Se debe a la alteración de la secreción de bilirrubina ya conjugada, desde el hepatocito al canalículo biliar, por enfermedad hepatocelular, y/o a una alteración del flujo biliar, por afectación del propio canalículo biliar o de la vía biliar extrahepática. En cualquiera de la dos situaciones, el acúmulo intrahepatocitario de bilirrubina conjugada y de otros compuestos que forman la bilis (ácidos biliares, conjugados, fosfolípidos, colesterol) resultante permite, por un lado, su paso a sangre y la consiguiente hiperbilirrubinemia conjugada (definida por una cifra de bilirrubina directa por encima de 2 mg/dl o su incremento por encima del 20% de la cifra total de bilirrubina en sangre) y, por otra, el acúmulo intra-celular con efecto tóxico sobre las propias células hepáticas y del epitelio biliar. Es esencial, a la hora de encarar el diagnóstico diferencial de una ictericia, la diferenciación entre hiperbilirrubinemia conjugada y no conjugada, ya que estas dos situaciones están ocasionadas por grupos de entidades muy diferentes. La ictericia por hiperbilirrubinemia conjugada (ictericia colestática) siempre es consecuencia de una enfermedad hepatobiliar, y su gravedad la determinan la severidad de la lesión hepática y de sus posibles consecuencias (insuficiencia o fallo hepático, hipertensión portal, encefalopatía, etc.). El tratamiento se basa en el tratamiento de la hepatopatía subyacente y en el tratamiento común del síndrome colestático (Tabla I).
Diagnóstico diferencial del niño con colestasis
Las posibles causas de ictericia por hiperbilirrubinemia conjugada son muy diferentes, según nos encontremos ante un recién nacido o lactante pequeño (menor de 6 meses) o ante un niño por encima de esa edad.
Ictericia en el lactante (colestasis del lactante)
La ictericia suele ser la forma clínica de debut de la hepatopatía en los lactantes. Debe ser descartada una colestasis, mediante la determinación de la cifra de bilirrubina directa, en todo recién nacido con ictericia prolongada (más de 15 dias). Es prioritario el diagnóstico precoz de aquellas entidades con posibilidad de tratamiento específico (atresia biliar, galactosemia, tirosinemia…).
Los recién nacidos y lactantes sanos presentan una “alteración fisiológica” del flujo biliar hasta completar la maduración anatómica y funcional del hígado en la vida post-natal. Esta inmadurez justifica que las enfermedades hepáticas que inciden en esta edad desencadenen una ictericia, como principal manifestación clínica, y de que patologías extrahepáticas (shock, sepsis) de suficiente gravedad puedan ir acompañadas de una colestasis en el recién nacido o lactante.
De cara al diagnóstico de un síndrome colestático en un recién nacido o lactante, es fundamental tener en cuenta que, en ocasiones, la ictericia colestática puede solaparse en el tiempo con una ictericia fisiológica del recién nacido, por lo que debe ser descartada una colestasis, mediante la determinación de la cifra de bilirrubina directa, en todo recién nacido con ictericia prolongada (más de 15 días).
La incidencia de colestasis neonatal se estima entre 1/2.500 y 1/5.000 recién nacidos. De forma general, la causa más frecuente, en niños con patología neonatal importante, sería la inmadurez hepática asociada a diferentes causas, como: prematuridad, cardiopatía, infección, cirugía, nutrición parenteral, etc. En el recién nacido o lactante sin patología neonatal, las causas más frecuentes son: en primer lugar, la atresia biliar extrahepática; y, en segundo lugar, el déficit de alfa-1-antitripsina, seguidas del síndrome de Alagille y de la colestasis intrahepática familiar progresiva (CIFP)(1-3).
El diagnóstico diferencial (ver algoritmo 1 y tabla II) de las distintas entidades puede ser complicado; ya que, a menudo, se solapan los rasgos clínicos, bioquímicos e histológicos. Se aconseja ingreso hospitalario para observación y diagnóstico más rápido, ya que es prioritario el diagnóstico precoz de aquellas entidades con posibilidad de tratamiento específico (atresia biliar, galactosemia, tirosinemia, panhipopituitarismo, etc.). Asimismo, será importante iniciar medidas para prevenir y tratar las consecuencias médicas y nutricionales del cuadro colestático (Tabla I).


En general, aunque alguna de las causas de colestasis del lactante sea susceptible de tratamiento eficaz, debe considerarse que no son procesos benignos, ya que muchas de ellas causan disfunción hepática crónica con alteración de la calidad de vida y/o necesidad de trasplante hepático.
Ictericia por inmadurez hepática asociada a patología neonatal grave
Es la causa más frecuente en niños con patología neonatal importante, por lo que se da fundamentalmente en un ámbito hospitalario. Entre las causas desencadenantes, destacan: inmadurez hepática (prematuridad), situaciones de hipoxia grave (cardiopatía, shock), infecciones (sepsis, infección urinaria), fracaso intestinal con necesidad de nutrición parenteral y escasos o nulos aportes enterales, infección connatal (TORCH) o infección neonatal (herpes simple, citomegalovirus, adenovirus…).
Habitualmente, existe relación entre la gravedad de la patología asociada y el grado de disfunción hepática y, en general, esta tiene buen pronóstico. La buena evolución del cuadro colestático, en paralelo a la resolución de sus factores precipitantes, confirma el diagnóstico. La base del tratamiento de este tipo de colestasis es el de la causa desencadenante.
Dentro de este grupo de colestasis secundarias, merecen ser comentadas las secundarias a un panhipopituitarismo congénito, puesto que presentan algunas características particulares dentro de las colestasis neonatales. La más típica es la presencia de hipoglucemia severa y de difícil control, acompañada de escaso desarrollo de caracteres sexuales (micropene) y, muchas veces, de alteraciones de línea media. Los hallazgos histológicos (lo más frecuente, una hepatitis neonatal por células gigantes con hipoplasia ductal) y las pruebas funcionales hormonales, junto con las de neuroimagen, confirman generalmente el diagnóstico. El tratamiento hormonal sustitutivo normaliza la función hepática(4,5).
Hepatopatías por trastorno hepato-biliar intrínseco (idiopático o genético)
Son las hepatopatías colestáticas más frecuentes en la edad infantil. Este grupo incluye:
Atresia biliar extrahepática: proceso inflamatorio progresivo que conduce a la obliteración de la vía biliar extrahepática, así como a la lesión del parénquima hepático (inflamación y fibrosis) y de la vía biliar intrahepática. Incidencia media de 1/10.000 RN vivos(6). Es la causa más frecuente de colestasis crónica y de trasplante hepático en la infancia. Etiología desconocida. Se reconocen dos formas clínicas: la embrionaria o sindrómica (10% de los casos), que asocia otras malformaciones (polisplenia, situs inverso, etc.), y la forma perinatal o adquirida (90% de los casos)(7), ambas con mecanismos patogénicos probablemente diferentes (defecto en la morfogénesis en la primera y una posible agresión viral o toxica postnatal con respuesta inmune en la segunda)(8). El cuadro clínico es muy característico, con un recién nacido a término, de peso y aspecto normal, que inicia ictericia con hipocolia entre las 2 y las 6 semanas de vida, con buen estado general, hepatomegalia firme, y posterior esplenomegalia. Analítica con signos de colestasis (aumento de bilirrubina total y directa, GGT mayor de 300 U/l), elevación moderada de transaminasas y coagulación normal.
El diagnóstico de atresia biliar debe ser considerado siempre ante un recién nacido con: ictericia, hepatomegalia y acolia. Lo apoya la ausencia de excreción intestinal de contraste en la gammagrafia hepática (HIDA), previa administración de fenobarbital y hallazgos compatibles en la biopsia hepática (colestasis, proliferación ductal, fibrosis portal) y lo confirma la laparotomía o laparoscopia exploradora. De hecho, esta debe ser realizada en todo RN con cuadro compatible o acolia mantenida y en el que no sea demostrada claramente otra etiología. El tratamiento es quirúrgico, con realización de una porto-entero-anastomosis (Kasai 1). De cara al restablecimiento del flujo biliar y consiguiente mejora del pronóstico, son fundamentales el diagnóstico y tratamiento precoces (antes de los dos meses de vida). El empleo de antibióticos tras la intervención es variable según los diferentes equipos; sin embargo, la mayoría de los grupos mantienen tratamiento inicial intravenoso y posteriormente profilaxis oral, para reducir el riesgo de colangitis, durante al menos un año, alternando diferentes antibióticos, como: amoxicilina, cefalexina y amoxicilina-clavulánico.
Sin tratamiento, la mortalidad es del 100% antes de los 3 años de vida, por desarrollo de cirrosis biliar e insuficiencia hepática. Tras la cirugía, un 30% no restablecerán flujo biliar y otro 20% lo harán de forma parcial, precisando todos ellos un trasplante hepático en los meses siguientes. Del 50% restante que restablece el flujo biliar tras la cirugía, un 70% de ellos precisará a largo plazo trasplante hepático por evolución cirrógena, ya que la lesión del parénquima persiste a pesar del buen resultado quirúrgico. La supervivencia a los dos, cuatro y diez años de vida, sin necesidad de trasplante, se situa alrededor del 55%, 45% y 35%, respectivamente(9,10).
Se han referido como factores predictivos de mala evolución post-Kasai: la forma embrionaria de la enfermedad, la existencia de fibrosis severa, la edad superior a los dos meses en el momento de la cirugía o la falta de experiencia tanto quirúrgica como en el manejo postoperatorio del centro.
El tratamiento médico post-cirugía se basa en un correcto soporte nutricional y en el tratamiento general de toda colestasis (Tabla I). El tratamiento con corticoides ha demostrado un efecto beneficioso tan solo en cuanto a la mejoría analítica, con descenso de las cifras de bilirrubina y transaminasas en sangre, pero no referente a la la reducción de la necesidad de trasplante, lo que cuestiona su utilidad(11).
Síndrome de Alagille: caracterizado por la asociación de un cuadro de colestasis con: escasez de conductos biliares intrahepáticos, alteraciones cardíacas (la más frecuente, una estenosis pulmonar periférica), alteraciones vertebrales (“vertebras en mariposa”), oculares (embriotoxon posterior) y facies peculiar con hipertelorismo, abombamiento frontal y mentón prominente. La incidencia estimada es de 1/70.000-100.000 recién nacidos vivos, aunque podría ser mayor, debido a que solo aquellos casos con afectación más severa son los reconocidos clínicamente. La enfermedad es debida a una mutación en el gen JAG1 del cromosoma 20 (90% de los casos) o en el gen NOTCH 2 en el cromosoma 1 (1% de los casos). La herencia es autosómica dominante con penetrancia completa (96%), pero con gran variabilidad en su expresión clínica (solo el 50-55% de los casos cumplen los criterios clínicos diagnósticos). En un 50-70%, son mutaciones “de novo”, no presentes en los progenitores. Frecuente afectación de hermanos, pero con la posibilidad de expresión clínica muy variable(12).
La colestasis es de inicio neonatal o en el lactante pequeño, con: ictericia, en el 80% de los casos, coluria, hipocolia, hepatomegalia y retraso en el desarrollo, con posterior aparición de prurito intenso y xantomas. Analítica con: hiperbilirrubinemia, elevación de ácidos biliares séricos, hipercolesterolemia y aumento de GGT y moderado de transaminasas. El diagnóstico lo da el cuadro clínico con asociación de, al menos, tres de los rasgos que definen el síndrome. En aquellos casos que no cumplen criterios clínicos, el estudio genético, con secuenciación de los genes implicados e identificación de la mutación causal, permite el diagnóstico. La biopsia hepática, con escasez de conductos biliares intrahepáticos, puede no ser evidente aún en el lactante pequeño(13).
No tiene tratamiento específico, basándose el manejo médico en el adecuado soporte nutricional y en el tratamiento común del síndrome colestático. En aquellos niños con colestasis precoz, predomina una evolución marcada por la mala calidad de vida (prurito muy intenso y difícil de controlar, mala situación nutricional) y hasta un 50% desarrollan una colestasis grave con cirrosis biliar o fibrosis portal severa, que precisará de un trasplante hepático antes de la edad adulta(14,15).
Colestasis intrahepática familiar progresiva (CIFP): grupo heterogéneo de enfermedades con base genética (herencia autosómica recesiva) y especial incidencia en determinados grupos étnicos. En el 50% de los casos, existe antecedente familiar o consanguinidad. Son trastornos diferentes, que tienen en común la alteración en la formación de bilis secundaria a mutaciones en genes de los sistemas de transporte canalicular de los hepatocitos, con resultado de ausencia o disfunción de las proteínas implicadas y el desarrollo de un cuadro de colestasis crónica de debut temprano, con prurito severo y mal pronóstico general con evolución cirrógena.
Se han identificado varios subtipos según la localización del defecto y la mutación causante(16):
• CIFP 1: defecto de FIC1. Mutación en el gen ATP8B1 (cromosoma 18q21).
El FIC1 no es un transportador de ácidos biliares, sino una proteína de membrana (ATPasa tipo P), que parece funcionar transfiriendo aminofosfolípidos de la capa externa a la interna de la membrana del canalículo biliar, por lo que su defecto condiciona la alteración estructural de dicha membrana y su susceptibilidad al daño mediado por los ácidos biliares. Otros tejidos, como: hígado, páncreas, estómago, intestino delgado y riñón, también lo expresan. Las mutaciones del gen ATP8B1 condicionan dos enfermedades con diferente evolución: una forma grave (CIFP 1) y otra más benigna, con episodios colestáticos intermitentes con intervalos asintomáticos, sin daño histológico permanente (CIBR o colestasis intrahepática benigna recurrente). Los casos de CIFP1 desarrollan una hepatopatía colestática severa con desarrollo de fibrosis intensa o cirrosis. Es frecuente la asociación de una diarrea crónica con esteatorrea. Todos los casos presentan un acusado retraso de talla y un prurito constante que condiciona una mala calidad de vida.
Desde el punto de vista bioquímico, es característica la normalidad en los valores de GGT. El diagnóstico de sospecha es clínico y lo confirma el estudio del gen ATP8B1 con detección de la mutación.
• CIFP 2: defecto de BSEP, transportador de sales biliares conjugadas con taurina o glicina a la bilis, lo que causa su acúmulo intracelular y daño hepático. Mutación gen ABCB11 (cromosoma 2q24.). Expresión clínica con: ictericia temprana, prurito, esteatorrea y retraso de talla. Bioquímicamente, presentan cifras normales de GGT y colesterol. Todo ello es similar a los casos con defecto de FIC1, pero existen diferencias entre ellos que pueden orientar hacia uno u otro grupo. Mientras los pacientes con FIC1 suelen presentar manifestaciones extrahepáticas, los niños con CIFP2 debutan con: transaminasas más altas, alfa-fetoproteína alta, lesión histológica con células gigantes multinucleadas y una evolución más rápida a cirrosis e insuficiencia hepática. Además, desarrollan frecuentemente colelitiasis (32%) y pueden desarrollar hepatocarcinoma temprano. La respuesta al tratamiento con ácido ursodeoxicólico es mejor en este grupo de pacientes. El diagnóstico se basa en la detección de la ausencia de BSEP, mediante técnicas de inmuno-histoquímica en el hígado y la confirmación genética de la mutación(17).
Dentro de este grupo, recientemente se han descrito 2 variantes fenotípicas según presenten o no la mutación genética D 482G. En los pacientes que no presentan esta mutación, la severidad clínica, analítica y las complicaciones son mayores y más tempranas, así como la más frecuente necesidad de trasplante(18).
• CIFP 3: defecto de MDR3 (transporte de fosfolípidos) por mutación del gen ABCB4 (cromosoma 7q21). La ausencia de fosfolípidos en bilis condiciona un defecto en la solubilidad de esta y la obstrucción de los canalículos biliares, con el consiguiente daño hepático. La enfermedad es variable según la mutación condicione una proteína truncada o con actividad residual. El espectro clínico es más variable, que el de los niños con CIFP 1 y 2. La mayoría de los casos, debutan con hepatoesplenomegalia y complicaciones de cirrosis a lo largo de la infancia (incluso adolescencia), y, solo un tercio de los casos, en el primer año de vida. Es infrecuente la presentación con colestasis neonatal y, generalmente, se inicia en el lactante. Presentan prurito y piel seca e hiperqueratósica. Bioquímicamente, cursan con: aumento de GGT, transaminasas, bilirrubina y colesterol normal. Es frecuente la presencia de colelitiasis y los hallazgos histológicos típicos son: la proliferación ductal e infiltrado inflamatorio con fibrosis y/o cirrosis. Dependiendo del grado de déficit de MDR 3, pueden desarrollar una cirrosis en el curso de la primera década, con descompensación brusca y necesidad de trasplante hepático.
El diagnóstico se realiza mediante inmuno-histoquímica, mediante la demostración de una ausencia de expresión de MDR3 en la biopsia hepática. En aquellos casos con déficit parcial y expresión inmuno-histoquímica prácticamente normal, el estudio genético, con detección de la mutación, corrobora el diagnóstico, aunque no siempre hay coincidencia con las mutaciones actualmente conocidas.
El tratamiento de los defectos de transportadores biliares se basa, además del común al resto de las colestasis (Tabla II), en la inducción del flujo biliar con ácido ursodeoxicólico a altas dosis (20-30 mg/kg/día). La respuesta es variable según el tipo de colestasis y en aquellos casos en que resulte ineficaz, la única alternativa es el trasplante hepático.
Colestasis por defectos en la síntesis de los ácidos biliares: una causa infrecuente de colestasis neonatal es el defecto de síntesis de ácidos biliares por déficit de alguno de los enzimas que intervienen en ella, como: 3βhidroxi-Δ5-C27esteroide- oxidoreductasa (mutaciones en gen HSD3β7), 4-3-oxosteroide-5 β-reductasa (gen AKR1D) o Oxysterol 7-α-hidrolasa (gen CYP7B1). Las consecuencias clínicas derivan de: la hepatotoxicidad por acúmulo de metabolitos intermedios, la reducción del flujo biliar y la alteración en la absorción intestinal de grasas y vitaminas liposolubles por la disminución de ácidos biliares en bilis.
Presentan ictericia colestática precoz y hepatopatía severa, que puede evolucionar a cirrosis. No presentan elevación de ácidos biliares en sangre y, por tanto, no existe prurito. El diagnóstico se realiza mediante la detección en orina del aumento global de excreción de ácidos biliares, con reducción de ácido cólico/quenodeoxicólico y aumento de derivados. En algunos de los defectos, el pronóstico mejora con tratamiento sustitutivo precoz con ácidos biliares primarios (cólico y quenodeoxicólico), pudiendo llegar a normalizar la función hepática; mientras que, en otros, el único tratamiento es el trasplante hepático.
Déficit de alfa-1-antitripsina: afecta a 1/2.000 RN vivos, de los cuales, un 10-20% desarrollarán una hepatopatía de grado variable durante la infancia. Una mutación en el gen Serpina 1 en el cromosoma 14, condiciona la producción de una alfa-1-antitripsina anómala con acúmulo intrahepatocitario, como posible mecanismo patogénico. De los diferentes fenotipos, el PiZZ es el que con más frecuencia asocia déficit marcado y enfermedad. En un 50% de los casos, la enfermedad hepática debuta como una colestasis en los primeros meses de vida, con hipocolia y sin coagulopatía. La sospecha diagnóstica la establece el hallazgo de una alfa-1-antitripsina baja en sangre (menos de 100 mg/dl) y la confirmación: la determinación de un fenotipo o genotipo compatible (PiZZ) por electroforesis y PCR en sangre, respectivamente; la demostración en la biopsia hepática de los acúmulos de alfa-1-AT; o la detección de la mutación, mediante secuenciación del gen (Serpina 1).
No existe tratamiento específico y evoluciona con desaparición de la ictericia, en la mayoría de los casos con debut colestático, manteniendo una elevación de transaminasas y observándose, posteriormente, aparición lenta de signos de hipertensión portal con fibrosis portal progresiva, hasta llegar a una situación de insuficiencia hepática a diferentes edades (adolescencia o preadolescencia). En estos, la reaparición de la ictericia en la evolución se considera un hecho de mal pronóstico. Otro grupo mantiene la ictericia, con evolución rápida a cirrosis e insuficiencia hepática en el primer año de vida y, en otro pequeño grupo de niños, la enfermedad debuta tardíamente, con hepatomegalia o hallazgo casual de alteración de la función hepática y sin antecedente de colestasis neonatal. Se considera que un 60-70% de los que debutan con colestasis neonatal, precisarán trasplante hepático antes de los 15 años. El trasplante hepático está indicado ante la aparición de complicaciones por hipertensión portal o de signos de insuficiencia hepatocelular(19).
Enfermedad aloinmune del hígado fetal (anteriormente llamada hemocromatosis neonatal): entidad muy poco frecuente y de patogenia desconocida, que debuta con un cuadro clínico de colestasis y fallo hepático de rápida instauración en las primeras semanas de vida. La hipótesis más aceptada es la de un trastorno aloinmune gestacional, frente a un antígeno hepático actualmente no definido y mediado por IgG materna, que atraviesa la placenta. La intensa lesión hepática condicionaría secundariamente la alteración del flujo de hierro transplacentario y su depósito en diferentes órganos. El diagnóstico de sospecha se basa en la presentación clínica descrita, junto con el hallazgo de cifras elevadas de ferritina y alto porcentaje de saturación de la transferrina en sangre, y la posibilidad de hermanos previos afectos. Se confirma mediante la demostración de los depósitos de hierro en hígado u otras posibles localizaciones, pero en la práctica, la biopsia es impracticable, lo que obliga a biopsiar otros tejidos de menos riesgo, como las glándulas de la submucosa oral; el diagnóstico puede realizarse, también, de forma indirecta, mediante RMN y estimación del depósito férrico aumentado en páncreas, suprarrenales, corazón, etc. Es imprescindible el tratamiento médico precoz, con exanguinotransfusión y gammaglobulina (1 g/kg iv), junto a las habituales medidas de soporte de fallo hepático. La supervivencia estimada con este tratamiento alcanza el 75% en alguna de las series publicadas, quedando la opción del trasplante hepático para aquellos casos en los que no se obtiene respuesta(20).
Enfermedades metabólicas: diferentes metabolopatías pueden producir una ictericia-colestasis de inicio precoz; si bien, esta no suele ser el síntoma predominante, sino otros hallazgos, como:
• Fallo o insuficiencia hepática, con déficit de la actividad de protrombina, que no se corrige tras administración de vitamina K, hipoglucemia y colinesterasa baja. Es el caso de la galactosemia, tirosinemia o fructosemia.
- Tirosinemia: error innato del metabolismo de la tirosina por déficit de fumarilacetoacetasa, ocasiona una enfermedad grave con: hepatopatía, disfunción tubular renal, con raquitismo hipofosfatémico, y un síndrome similar a la porfiria. Herencia autosómica recesiva. Incidencia de 1 /100.000 RN. En el 77% de los afectos, la enfermedad se manifiesta en los primeros 6 meses de vida como una hepatopatía grave o insuficiencia hepática (forma aguda). En un 14%, los síntomas aparecen después del 6º mes de vida con: retraso ponderal, raquitismo, hepatomegalia y coagulopatía (forma subaguda). Un 9% de los casos consulta pasado el primer año de vida por hepatomegalia, raquitismo y retraso en el crecimiento (forma crónica). Todos ellos presentan un riesgo elevado de desarrollar cáncer de hígado, con una incidencia de hepatocarcinoma del 40% a los 3 años de edad.
El diagnóstico se realiza tras el hallazgo patognomónico de succinil-acetona, en orina o en sangre, o mediante comprobación del déficit de fumarilacetoacetasa en fibroblastos de piel. La elevación sérica de tirosina o fenilalanina no son específicas de la enfermedad.
Se ha relacionado el pronóstico con la edad de debut de la enfermedad. Así, la supervivencia a los dos años del debut fue del 29% en aquellos sintomáticos antes de los 2 meses de edad, frente a un 96% que debutaron por encima de los 6 meses de edad. Actualmente, el tratamiento de elección es la nitisinona (NTBC), que mediante bloqueo enzimático reduce la producción de los metabolitos tóxicos patogénicos, asociada a la restricción dietética de tirosina y fenilalanina. El trasplante hepático quedaría reservado a aquellos casos que, excepcionalmente, no respondan al tratamiento médico o diagnosticados en estadios tardíos o con hepatocarcinoma.
- Otras: Fructosemia: intolerancia hereditaria a la fructosa por déficit de fructosa-1-P-aldolasa. Se manifiesta como hepatopatía grave con signos de insuficiencia hepática y tubulopatía renal al poco tiempo de ser iniciada la ingesta de fructosa por el niño. Característico el rechazo de alimentos que contengan fructosa. La confirmación diagnóstica se realiza mediante cuantificación de la actividad enzimática en tejido hepático o mucosa intestinal. El tratamiento consiste en la exclusión completa de la dieta de fructosa, sacarosa y sorbitol. Galactosemia: trastorno autosómico recesivo que produce una intolerancia a la galactosa por déficit de galactosa 1-fosfato-uridiltransferasa. La presentación clínica puede variar desde la instauración de un fallo hepático, siguiendo las primeras tomas de leche, hasta formas menos agudas (vómitos, diarrea, ictericia…) de inicio en los primeros días de vida. La sospecha se sustenta en la detección de cuerpos reductores en orina y la confirmación se establece tras la determinación de la actividad enzimática en eritrocitos. El tratamiento consiste en la exclusión de la lactosa de la dieta.
• Hepatomegalia. Glucogenosis I y III, Lipidosis (enfermedad de Nieman-Pick A y C), enfermedad de Wolman (déficit de lipasa ácida).
El tratamiento médico de sostén y el apoyo nutricional es común a todas las entidades que cursan con colestasis crónica. Se basa en el uso de fármacos favorecedores del flujo biliar (fenobarbital, ácido ursodeoxicólico), vitaminas liposolubles, extractos pancreáticos, así como en la prevención y tratamiento de las infecciones intercurrentes y las complicaciones derivadas de la afectación de la función hepática (Tabla I).
Colestasis en el lactante y niño mayores
En este grupo de edad, el espectro de causas de un síndrome colestático varía significativamente respecto al lactante menor de 3-6 meses, lo que modifica el proceso diagnóstico diferencial (Algoritmo 2).
Colestasis secundaria a lesión hepatocelular
Característicamente, predominan los signos de lesión hepatocitaria, como la elevación de transaminasas, o si la lesión hepática es de suficiente severidad, los signos y síntomas de insuficiencia hepatocelular, como hipoalbuminemia o coagulopatía que no responde a vitamina K, sobre otros signos más específicos de afectación biliar, como elevación de GGT o de fosfatasa alcalina. Teniendo en cuenta que, las posibles causas de hepatopatía en el niño son múltiples y todas ellas pueden producir ictericia en un momento dado de su evolución, nos referiremos a aquellas entidades que con mayor frecuencia la presentan. Aunque en la mayoría de estas enfermedades (p. ej.: enfermedad de Wilson, hepatitis autoinmune, etc.) no es la ictericia la forma más común de presentación, siempre hay que tenerlas en cuenta en el diagnóstico diferencial de una ictericia colestática.
Hepatitis viral aguda. (Ver capítulos correspondientes en este mismo número de la revista).
Hepatitis por fármacos. Numerosos fármacos pueden producir una hepatitis ictérica mediada, bien por un mecanismo de toxicidad directa o por una reacción de idiosincrasia. Entre los descritos con mayor frecuencia, se encuentran:
• Paracetamol o acetaminofén: su toxicidad hepática, con aparición de ictericia o desarrollo de un fallo hepático fulminante, puede aparecer, bien tras la ingesta de una única dosis o tras 2-3 días de continuada ingesta de dosis altas (20-30 mg/kg). La dosis letal estimada en adultos es de 140 mg/kg de peso. Se ha descrito la menor incidencia de hepatotoxicidad tras intoxicación por paracetamol en niños menores de 5 años (menos del 5,5%) frente a adolescentes (29%) o adultos con similares niveles tóxicos en sangre. El tratamiento puede implicar la utilización de la n-acetil-cisteína como antidoto, hemodialisis precoz en casos seleccionados, medidas de soporte de fallo hepático e incluso trasplante hepático en los casos refractarios a estas medidas.
• Otros: isoniacida, valproato sódico, etc.
Hepatitis autoinmune. Enfermedad caracterizada por una hepatopatía, generalmente grave, asociada con una serie de autoanticuerpos circulantes. El daño hepático es mediado por una respuesta inflamatoria dirigida frente a componentes de los hepatocitos. No se ha podido delimitar con exactitud el papel de los autoanticuerpos detectados en dicha respuesta inflamatoria. La etiología es desconocida, aceptándose la idea de que, sobre una determinada predisposición genética, determinados factores exógenos (virales, fármacos) podrían disparar la enfermedad. Se clasifica según el tipo de autoanticuerpos circulantes presentes en: Tipo 1: antinucleares (ANA) y anti músculo liso (SMA) anti-actina positivos; Tipo 2A: anti LKM (anticuerpos amtimicrosomales de hígado y riñón); Tipo 2B: anti-LKM asociado a infección crónica por el virus de la hepatitis C; y Tipo 3: anti-antígeno soluble hepático (SLA).
El debut de la enfermedad puede ser muy heterogéneo, desde una forma similar a una hepatitis aguda, con: ictericia, precedida de anorexia, dolor abdominal o malestar general (más del 50% de los casos), a un fallo hepático fulminante (10-15% de los casos, más frecuente en tipo anti-LKM), o un debut más insidioso con síntomas inespecíficos (anorexia), hepatomegalia firme u otros estigmas de hepatopatía crónica. En un 20%, asocian otros trastornos autoinmunes (tiroiditis, enfermedad inflamatoria intestinal, diabetes mellitus, anemia hemolítica, etc.). El diagnóstico exige el cumplimiento de los siguientes criterios:
• Biopsia hepática con signos de hepatopatía crónica con: necrosis en sacabocados, hepatitis lobular o necrosis en puentes, y sin otros hallazgos que sugieran otra etiología.
• Hipergammaglobulinemia.
• Seropositividad (títulos por encima de 1:30) para autoanticuerpos ANA, Anti-LKM o SMA.
• Despistaje negativo para otras causas de hepatopatía. En la analítica, siempre existe una elevación de transaminasas de diferente rango con característica escasa elevación de fosfatasa alcalina y GGT. Predominan las niñas (80%) y la edad media al diagnóstico oscila entre los 7-10 años, aunque también se han diagnosticado por debajo del año de vida.
El tratamiento debe intentarse en todos los casos y consta de corticoides (prednisolona a 2 mg/kg/día con un máximo de 60 mg/día) y azatioprina (2 mg/kg/día). Como alternativa terapéutica, se ha introducido el empleo de ciclosporina. Debe considerarse el trasplante hepático en casos de: fallo hepático fulminante, cirrosis descompensada o tratamiento médico fallido o con efectos secundarios intolerables(21).
Colangitis esclerosante primaria. Trastorno inflamatorio crónico de la vía biliar intra y extra-hepática, que condiciona el desarrollo de una colestasis y fibrosis hepática progresiva con hipertensión portal. Etiología probablemente autoinmune, sugerida por la presencia, en muchos de los casos, de marcadores de autoinmunidad (dos tercios de los casos presentan positividad para: anticuerpos anti-nucleares, anti-músculo liso o hipergammaglobulinemia) y, en ocasiones, hallazgos histológicos característicos de hepatitis autoinmune (considerándose síndromes de superposición entre ambas entidades). Es habitual la asociación con enfermedades extra-hepáticas, la más frecuente de las cuales es la enfermedad inflamatoria intestinal (en particular, la colitis ulcerosa). La edad de comienzo es muy variable, incluso puede ser neonatal, y su naturaleza insidiosa puede retrasar el diagnóstico. Presenta muy diversas formas de debut clínico: ictericia, dolor abdominal, pérdida de peso, colangitis con prurito, complicaciones derivadas del desarrollo de cirrosis o detección accidental de hepatomegalia o disfunción hepática. El diagnóstico se basa en la visualización, mediante colangiografia o colangio-resonancia, de la imagen característica de una vía biliar intrahepática arrosariada, con dilataciones saculares, estenosis múltiples y una porción extra-hepática muy irregular. La lesión histológica típica consiste en una colangitis con fibrosis obliterativa de los ductos biliares. El tratamiento médico incluye: ácido ursodeoxicólico (15 mg/kg/día), que puede mejorar el prurito y la función hepática, y el tratamiento precoz de las colangitis con antibióticos, además del tratamiento común al resto de cuadros colestáticos. Los inmunosupresores no son tan efectivos como en la hepatitis autoinmune, salvo que se trate de un cuadro de superposición con esta. Debe ser evitada, siempre que sea posible, la cirugía biliar por su, generalmente, poca influencia positiva en la progresión de la enfermedad y su potencial aumento de la dificultad en un eventual trasplante hepático, único tratamiento posible en aquellos casos con mala evolución(21).
Hepatopatías de base metabólica. A diferencia del recién nacido, en etapas posteriores, la ictericia no es una forma habitual de presentación de las enfermedades metabólicas, siendo más frecuentes otras, como: hepatomegalia, esplenomegalia, alteraciones neurológicas o del crecimiento, etc.
Disfunción hepática asociada a fracaso intestinal y nutrición parenteral. Los pacientes con fracaso intestinal y nutrición parenteral prolongada pueden desarrollar una hepatopatía de variable intensidad, desde una esteatosis hepática hasta una cirrosis terminal. Se han implicado varios factores en su desarrollo (toxicidad de elementos de las fórmulas parenterales con especial efecto negativo de los fitosteroles y ácidos grasos poli-insaturados w-6) bacteriemias de origen en el cateter central, ausencia de tránsito intestinal, etc.), aceptándose en este momento una etiología multifactorial como la más probable. El momento de la aparición del cuadro colestático es muy variable de unos pacientes a otros y pueden asociar signos-síntomas de hipertensión portal (esplenomegalia, hiperesplenismo, ascitis, etc.). El diagnóstico exige descartar otras posibles causas de colestasis en este tipo de pacientes.
La hepatopatía es reversible si se suspende la nutrición parenteral antes del desarrollo de fibrosis severa o cirrosis. Si esta suspensión no es posible, el tratamiento va enfocado a la prevención de factores precipitantes: la infección (cuidados del catéter central, descontaminación oral), estasis intestinal (introducción de aportes enterales si es posible), estasis biliar (ácido ursodeoxicólico oral a dosis de 10-20 mg/kg/día) y la adecuación constante de la composición de la parenteral a la situación del paciente (fórmula lipídica con una proporción favorable de ácidos grasos w-6: w-3, aporte adecuado de proteínas, oligoelementos, adicción de glutamina, etc.). Como todo cuadro colestático, precisan un correcto aporte de vitaminas hidro y liposolubles(22).
Fibrosis quística. La prevalencia de la hepatopatía, definida esta como: un espectro de manifestaciones, desde hepatomegalia, alteración funcional asintomática o alteraciones ecográficas compatibles, hasta una cirrosis terminal asociada a esta enfermedad, se estima entre un 17-38%. En los casos de desarrollo de hepatopatía grave, cirrosis multilobular (5-10% de todos los pacientes con FQ), predominan los signos-síntomas derivados de hipertensión portal frente a la ictericia colestática. Otras posibles causas de ictericia son: casos con colestasis neonatal (menos de 2%), colelitiasis (1-27%), colangitis esclerosante (menos del 1%) o estenosis del colédoco (menos de 2%). Se ha asociado el desarrollo de hepatopatía severa con el antecedente de íleo meconial neonatal o los casos con insuficiencia pancreática. Habitualmente, la hepatopatía comienza en la infancia con un pico de incidencia en la adolescencia y caída posterior. El objetivo del tratamiento se basa en minimizar el daño hepático y la progresión a cirrosis, en el tratamiento habitual de la colestasis y en el manejo de posibles complicaciones de la hipertensión portal. El ácido ursodeoxicólico (15-20 mg/kg/día) mejora los parámetros bioquímicos de disfunción hepática, pero no se ha demostrado que evite la evolución a cirrosis. El trasplante hepático está indicado en los casos con hipertensión portal severa o desarrollo de insuficiencia hepática sin contraindicación pulmonar(23).
Enfermedad de Wilson. La enfermedad de Wilson puede justificar disfunción hepática, tan pronto, como a los 2 años de edad. Es un trastorno hereditario del metabolismo del cobre que se transmite a través de un gen recesivo localizado en el cromosoma 13. Se desconoce el defecto básico, pero se acepta el acúmulo de cobre en hígado y otros tejidos como el mecanismo responsable de las consecuencias clínico-patológicas de la enfermedad. En niños, predomina la afectación hepática (95%) frente a otras posibles manifestaciones (SNC, etc.). En la mayoría de los casos pediátricos, los pacientes se encuentran asintomáticos o con síntomas inespecíficos en el momento del diagnóstico. Poco frecuente, es la presentación con síntomas, como: ictericia, hepatoesplenomegalia o ascitis. Para el diagnóstico, se utilizan como cribado la determinación de la cifra de ceruloplasmina en sangre (reducida en un 88% de los casos) y de la excreción urinaria de cobre (incrementada en un 81%), pero el diagnóstico definitivo lo da el hallazgo de exceso de cobre en tejido hepático (por encima de 250 µg/g de tejido seco) y el estudio genético (mutaciones del gen ATP7B). El tratamiento se realiza con quelantes del cobre (penicilamina, trientine) o inhibidores de su absorción intestinal (acetato de zinc), que son eficaces en impedir la progresión de las lesiones dependiendo de la elección entre ambos tipos de fármacos, de la situación clínica de la enfermedad en un paciente concreto. El trasplante hepático está indicado en aquellos casos, que debutan como un fallo hepático fulminante o en los refractarios al tratamiento médico.
Lo ideal es su diagnóstico antes de que exista sintomatología hepática, pues la capacidad de respuesta al tratamiento con penicilamina o sulfato de zinc depende de ello. Los niños diagnosticados al detectarse elevación de AST/ALT, sin hiperbilirrubinemia ni descompensación ascítica, resuelven en un plazo inferior a 2 años la alteración funcional bajo tratamiento y se impide la evolución natural a cirrosis o daño neurológico(24).
Ictericia secundaria a afectación de la vía biliar
Quiste de colédoco. Anomalía más frecuente de la vía biliar extrahepática después de la atresia biliar. Incidencia estimada en países occidentales de 1/100.000 recién nacidos vivos. Etiología incierta con varias hipótesis (recanalización desigual de los conductos biliares durante la embriogénesis, regurgitación de enzimas pancreáticos al conducto biliar común por una unión anómala pancreático-biliar, etc.). Se distinguen tres formas: dilatación quística del colédoco, divertículo congénito del colédoco y coledocele congénito. El comienzo de los síntomas aparece a cualquier edad. En período de lactante, recuerdan a una atresia biliar, con ictericia prolongada, acolia y coluria. En niños mayores, no siempre hay ictericia y son frecuentes el dolor cólico epigástrico, las náuseas y los vómitos. La evolución sin tratamiento es hacia una cirrosis biliar. El diagnóstico se hace mediante ecografía, TAC o RMN abdominal. El tratamiento es quirúrgico, con escisión completa del quiste colédoco-yeyunostomía. En los casos de debut precoz con un cuadro de colestasis, el tratamiento quirúrgico debe ser precoz debido al rápido desarrollo de fibrosis hepática. En los casos de diagnóstico tardío y consiguiente evolución a cirrosis biliar, la única alternativa de tratamiento es el trasplante hepático.
Colecistitis calculosa o acalculosa. (Ver capítulo correspondiente incluido en este mismo número)
Tumores de la vía biliar. El más característico es el rabdomiosarcoma, que, aunque extremadamente raro, aparece casi exclusivamente durante la infancia (con frecuencia menor de 5 años). Debut clínico con: ictericia, hepatomegalia, coluria e hipocolia. Pobre supervivencia a pesar de resección, quimioterapia y radioterapia. Otros tumores pueden producir una compresión extrínseca de la vía biliar (linfomas, etc.), con aparición de un cuadro de ictericia obstructiva.
Función del pediatra en Atención Primaria
La función del pediatra de Atención Primaria, en referencia al síndrome colestático, se concreta en dos aspectos. El primero de ellos, es el de la detección-diagnóstico de una colestasis. En una gran mayoría de ocasiones, será la consulta de Atención Primaria donde acuda el niño con colestasis para una primera valoración. Identificar la ictericia y otros signos característicos de una colestasis, como son: la coluria, la hipo-acolia o el prurito, es el paso decisivo para el diagnóstico. Mayor trascendencia adquiere dicha detección, en el caso del recién nacido o lactante menor de dos meses con ictericia; ya que, en ocasiones, el diagnóstico precoz de entidades susceptibles de tratamiento, como es el caso de la atresia biliar extrahepática, influye significativamente en el pronóstico. Así, es obligatoria la realización de una determinación de la cifra en sangre de bilirrubina conjugada, en todo aquel lactante con una ictericia de más de dos semanas de evolución. Una vez detectada la colestasis, el gran número de posibles etiologías y la complejidad diagnóstica de algunas de ellas, requiere la consulta en un centro especializado.
El segundo aspecto alude al seguimiento post-diagnóstico. Muchas de las colestasis infantiles son procesos crónicos que, además de un seguimiento en un centro especializado más focalizado en la enfermedad hepática, requerirán un control cercano por parte del pediatra en aquellos aspectos no directamente relacionados con la hepatopatía (vacunaciones, desarrollo póndero-estatural, enfermedades intercurrentes etc.).
Bibliografía
Los asteriscos reflejan el interés del artículo a juicio del autor.
1. Mieli-Vergani G, Howard ER, Portman B, Mowat AP. Late referral for biliary atresia, missed opportunities for efective surgery. Lancet. 1989; 1: 421-3.
2. Stormon MO, Doney SF, Kamath KR, et al. The changing pattern of diagnosis of infantile cholestasis. J Paediatr Child Health. 2001; 37: 47-50.
3.*** Roberts E. The jaundiced baby. En Kelly DA ed. Diseases of the liver and biliary system in children. Ed Blackwell Publishing. 2004; p. 35-73.
4. Torberson M, Hart J, Westerhoff M el al. Neonatal giant cell hepatitis: Histological and etiological finding. Am J Surg Pathol. 2010; 34: 1498-503.
5. Binder G, Martin DD, Kanther I, et al. The course of neonatal cholestasis in congenital combined pituitary hormone deficiency. J Pediatr Metabol. 2007; 20: 695-702.
6. Stringer MD and Howard ER. Surgical disorders of the liver and bile ducts and portal hypertension. En: Kelly Deirdre A ed. Diseases of the liver and biliary system in children. 2nd ed. Blackwell Publishing. 2004. p324-62.
7. Sokol R, Shepherd R, Superina R, Bezerra J, Robuck P and Hoofnagle J. Screening and outcomes in biliary atresia: Summary of a National Institutes of Health Workshop. Hepatology. 2007; 46: 566-81.
8. Sokol RJ and Mack C. Etiopathogenesis of biliary atresia. Semin Liver Dis. 2001; 21: 517-24.
9. Kelly D, Davenport M. Current management of biliary atresia. Arch Dis Child. 2007; 92: 1132-5.
10. Chardot C and Serinet MO. Prognosis of biliary atresia: what can be further improved? Editorial. J Pediatr. 2006; 148: 432-5.
11. Davenport M, Parsons C, Tizzard S, Hadzic N. Steroids in biliary atresia: single surgeon, single centre, prospective study. J Hepatol. 2013; 59(5).
12. Warthen DM, Moore EC, Kamath BM, Morrissette JJ, Sanchez P, Piccoli DA, Krantz ID, Spinner NB Jagged1 (JAG1) mutations in Alagille syndrome: increasing the mutation detection rate. Hum Mutat. 2006; 27: 436-43.
13. Emerick KM, Rand EB, Goldmuntz E, Krantz ID, Spinner NB, Piccoli DA. Features of Alagille syndrome in 92 patients: frequency and relation to prognosis. Hepatology. 1999; 29: 822-9.
14. Englert C, Grabhorn E, Burdelski M, Ganschow R. Liver transplantation in children with Alagille syndrome: indications and outcome. Pediatr Transplant. 2006; 10: 154-8.
15. Lykavieris P, Hadchouel M, Chardot C, Bernard O. Outcome of liver disease in children with Alagille syndrome: a study of 163 patients. Gut. 2001; 49: 431-35.
16.*** L. Hierro, P. Jara. Colestasis infantiles y transportadores biliares. Gastroenterol Hepatol. 2005; 28(7): 388-95.
17.*** Pawlikowska L, Strautnieks S, Jankowska I, et al: Differences in presentation and progression between severe FIC1 and BSEP deficiencies. J Hepatol. 2010; 53: 170-8.
18. Davit-Spraul A, Fabre M, Branchereau S, et al: ATP8B1 and ABCB11 Analisis in 62 children with normal gamma-glutamyl transferasa progressive familial intrahepatic cholestasis (PFIC): Phenotypic differences between PFIC1 and PFIC2 and natural history. Hepatology. 2010; 51: 1645-55.
19.*** Sveger T. The natural history of liver disease in alpha-1-antitrypsin deficient children. Acta Paediatr Scand. 1988; 77: 847-51.
20. Whitington PF1. Gestational alloimmune liver disease and neonatal hemochromatosis. Semin Liver Dis. 2012; 32: 325-32.
21. Mieli-Vergani G, Vergani D. Autoimmune paediatric liver disease. World J Gastroenterol. 2008; 14: 3360-7.
22. Kelly DA. Preventing parenteral nutrition liver disease. Early Hum Dev. 2010; 86(11): 683-7.
23. Lamireau T, Monnereau S, Martin S, Marcotte JE, Winnock M, Alvarez F. Epidemiology of liver disease in cystic fibrosis: a longitudinal study. J Hepatol. 2004; 41: 1041-4.
24. Nicastro E, Ranucci G, Vajro P, Vegnente A, Iorio R. Re-evaluation of the diagnostic criteria for Wilson disease in children with mild liver disease. Hepatology. 2010; 52: 1948-56.
Bibliografía recomendada
- Roberts E. The jaundiced baby. En Kelly DA ed. Diseases of the liver and biliary system in children. Ed Blackwell Publishing. 2004; p. 35-73.
Revisión de la etiología, diagnóstico diferencial, tratamiento de la ictericia colestática en el recién nacido y lactante.
- L. Hierro, P. Jara. Colestasis infantiles y transportadores biliares. Gastroenterol Hepatol. 2005; 28: 388-95.
Revisión actualizada del grupo de enfermedades encuadradas dentro del término de colestasis intrahepáticas familiares, con revisión de su etiología, presentación clínica, posibilidades de diagnóstico y tratamiento.
- Pawlikowska L, Strautnieks S, Jankowska I, et al: Differences in presentation and progression between severe FIC1 and BSEP deficiencies. J Hepatol. 2010; 53: 170-8.
Estudio multicéntrico en el que comparan, en un numeroso grupo de pacientes, las diferencias de presentación clínica, analítica y evolutiva de pacientes con colestasis por FIC1 y deficiencia de BSEP.
- Sveger T. The natural history of liver disease in alpha-1-antitrypsin deficient children. Acta Paediatr Scand. 1988; 77: 847-51.
Artículo clave para el conocimiento de la incidencia e historia natural de esta enfermedad en la infancia tras el seguimiento durante años de una amplia cohorte de niños diagnosticados al nacimiento, mediante despistaje de la enfermedad en todos los recién nacidos.
| Caso clínico |
|
Consulta de una niña de dos meses y medio, con colestasis desde periodo neonatal. Padres jóvenes sanos y hermana de 8 años sana. No consanguinidad. Embarazo normal. Parto a término. Peso: 3.640 g. Desde el 5º día de vida, presenta colestasis con coluria e hipocolia. A los dos meses, presenta ITU por E. Coli, que ha recibido tratamiento antibiótico, con controles posteriores negativos. Lactancia materna hasta el 4º mes. A los 6 meses, inicia prurito, bilirrubina T/D: 5,3/3,6 mg/dl con GGT “normal” (95) y transaminasas elevadas (AST/ALT: 320/254). Hemograma y coagulación normales. Hipovitaminosis severa. Exploración física BEG, bien nutrida, ictericia. Ptosis OI y tortícolis leve congénita. Abdomen: hepatomegalia a 3 cm de consistencia normal, no esplenomegalia. No ascitis. No hernias. AC y P: normal. Pruebas complementarias Analitica: BILIT/D: 7,9/4,3. GOT/GPT: 135/95. GGT: 95. Alfafetoproteína elevada en tres ocasiones (hasta 129.000 a los dos meses y medio). Infección connatal (–). Alfa-1antitripsina: Normal. Ecografía abdominal: hepatomegalia leve homogênea. Resto normal. HIDA con Tc-99: captación hepática normal con excreción enlentecida del isótopo a intestino. Histologia: colestasis. Transformación giganto-celular. Pequeños focos de inmadurez y hematopoyesis.
|


