|
| Temas de FC |
A. Carcavilla Urquí![]()
Coordinador de la Unidad Multidisciplinar de Rasopatías. Servicio de Endocrinología Pediátrica. Hospital Universitario La Paz. Madrid
| Resumen
Las RASopatías comprenden un grupo de trastornos clínicamente solapados causados por variantes genéticas que afectan a componentes de la cascada de señalización intracelular de las RAS-MAPKinasas. Aunque varios de estos trastornos son muy infrecuentes, juntos representan uno de los mayores grupos de síndromes con afectación multiorgánica conocidos. El síndrome de Noonan es el más frecuente de todos, y constituye una RASopatía prototípica de la que el resto de RASopatías, como el síndrome cardiofaciocutáneo, el síndrome de Costello, o el síndrome de Noonan con lentiginosis múltiple, se diferencian tanto clínica como genéticamente. Aunque el diagnóstico es clínico, el estudio molecular es una herramienta inestimable en el diagnóstico y evaluación pronóstica. El tratamiento en el síndrome de Noonan es fundamentalmente sintomático, ya que no existen tratamientos etiológicos disponibles; sin embargo, el conocimiento de la base molecular de estos trastornos abre la vía para la investigación de tratamientos dirigidos a la raíz de este grupo de enfermedades. |
| Abstract
RASopathies comprise a group of clinically overlapping disorders caused by genetic variations affecting components of the RAS-MAPKinases intracellular signaling cascade. Although several of these disorders are very rare, together they represent one of the largest known groups of syndromes with multiorgan involvement. Noonan syndrome is the most frequent of all, and constitutes a prototypical RASopathy from which the other RASopathies, such as cardiofaciocutaneous syndrome, Costello syndrome, or Noonan syndrome with multiple lentiginosis, differ both clinically and genetically. Although the diagnosis is clinical, the molecular study is an invaluable tool in the diagnosis and prognostic evaluation. Treatment in Noonan syndrome is primarily symptomatic, as there are no etiologic treatments available; however, knowledge of the molecular basis of these disorders opens the way for research into treatments directed at the root cause of the associated problems. |
Palabras clave: Síndrome de Noonan; Síndrome cardiofaciocutáneo; Síndrome de Costello; Vía RAS/MAPK.
Key words: Noonan syndrome; Cardiofaciocutaneous syndrome; Costello syndrome; RAS/MAPK pathway.
Pediatr Integral 2024; XXVIII (5): 319 – 325
OBJETIVOS
• Conocer la base fisiopatológica común al grupo de las RASopatías.
• Identificar las manifestaciones clínicas características del síndrome de Noonan.
• Conocer los criterios clínicos que permiten diagnosticar el síndrome de Noonan, así como sus causas genéticas y los elementos de correlación genotipo-fenotipo.
• Conocer las particularidades clínicas y moleculares que distinguen a cada una de las RASopatías del síndrome de Noonan, así como el diagnóstico diferencial con otras entidades.
• Familiarizarse con los tratamientos disponibles en el síndrome de Noonan y las recomendaciones de seguimiento actuales.
Síndrome de Noonan y otras RASopatías
Introducción
El síndrome de Noonan y las RASopatías son trastornos genéticos con solapamiento clínico debidos a alteraciones en la regulación de la vía de las RAS-MAPKinasas.
El síndrome de Noonan es un trastorno multisistémico de origen genético y herencia autosómica dominante. Caracterizado por la tríada de fenotipo facial característico, cardiopatía congénita y talla baja; puede presentar además, en grado variable, problemas de alimentación, anomalías linfáticas, manifestaciones musculoesqueléticas, anomalías genitourinarias, trastornos del neurodesarrollo, tendencia al sangrado y predisposición a neoplasias, entre otras manifestaciones(1). En las últimas décadas se ha producido un desarrollo notable del conocimiento de su causa genética y su sustrato bioquímico, que reside en una alteración en la regulación de la vía de señalización intracelular de las RAS-MAPKinasas(2). Se han descrito diversos síndromes solapados clínicamente con el síndrome de Noonan, como el síndrome cardiofaciocutáneo, el síndrome de Costello, el síndrome de Noonan con lentiginosis múltiple (anteriormente denominado síndrome LEOPARD) o el síndrome de Noonan-like con cabello anágeno suelto, los cuáles se deben también a alteraciones en la regulación de la vía de las RAS-MAPKinasas; por ese motivo, se ha denominado colectivamente a ese grupo de síndromes RASopatías(2). La neurofibromatosis tipo 1 también se debe a una alteración de la regulación de las MAPKinasas, y es considerada igualmente una RASopatía.
Epidemiología
El síndrome de Noonan tiene una incidencia estimada de 1:1.000-2.500, y la neurofibromatosis tipo 1 de 1:2.500-3.000, mientras que el resto de RASopatías son bastante menos frecuentes.
El síndrome de Noonan es un trastorno relativamente frecuente, con una incidencia que se estima en uno por cada 1.000 a 2.500 recién nacidos vivos. Sin embargo, su incidencia real es desconocida y, dada su extrema variabilidad clínica, puede pasar sin diagnosticar, tanto en casos leves(3) como en casos de letalidad neonatal(4).
La neurofibromatosis tipo 1 tiene una incidencia estimada de 1 de cada 2.500-3.000, y el resto de RASopatías son mucho menos frecuentes, con incidencias estimadas de 1 de cada 100.000 para el síndrome de Noonan con lentiginosis múltiple(1), 1 de cada 800.000 para el síndrome cardiofaciocutáneo, y 1 de cada 1.290.000 para el síndrome de Costello(5). El síndrome de Noonan es la segunda causa más frecuente de cardiopatía congénita sindrómica y, consideradas en conjunto, las rasopatías son uno de los trastornos del desarrollo más frecuentes, con una incidencia acumulada de 1 de cada 1.000 recién nacidos.
Etiología y fisiopatología
Las RASopatías comparten una patogénesis molecular común, en la que las variantes genéticas afectan a componentes o moduladores de la cascada de las RAS-MAPKinasas, lo que conduce a un aumento del flujo de señal a través de esta vía.
En el año 2001 se identificó el primer gen relacionado con el síndrome de Noonan mediante una técnica de búsqueda de candidatos por posicionamiento: PTPN11. En este estudio, los autores identificaron que las variantes en PTPN11 eran la causa del síndrome de Noonan en cerca de un 50 % de los pacientes(6). PTPN11 codifica para SHP2, una protein-tirosín-fosfatasa involucrada en la vía de las RAS-MAPKinasas, una vía de señalización intracelular implicada en múltiples procesos celulares. Las mutaciones en PTPN11, de ganancia de función, producen un aumento de la actividad de SHP2, que en último término conducen a una hiperactivación de la vía RAS. A partir de este hallazgo, comenzó una etapa de búsqueda de genes mediante búsqueda de candidatos funcionales, que progresivamente involucró a otros genes de la vía de las RAS-MAPkinasas en la etiología del síndrome de Noonan y las otras RASopatías. En los últimos 10 años, el desarrollo de técnicas de secuenciación genómica ha permitido la búsqueda “libre de hipótesis” de nuevos genes. Como consecuencia de estos desarrollos, en la actualidad, se han descrito más de 20 genes asociados a las RASopatías. De cualquier manera, la heterogeneidad de locus para algunos síndromes, junto con la heterogeneidad de alelo de otros, hace la clasificación de las RASopatías más compleja(2).
Dada la expansión de las técnicas de secuenciación genómica y el número creciente de genes documentados en las RASopatías, es imperativo contar con estrategias para validar estos hallazgos. En esa línea, el panel de expertos en rasopatías de la Clinical Genome Resource (ClinGen), fundada por los National Institutes of Health, ha publicado una revisión de 19 genes implicados en rasopatías, que aporta una clasificación de la fuerza de la evidencia clínica y experimental entre los distintos genes y los fenotipos a los que se han asociado(7). Se han descrito múltiples ejemplos de correlación genotipo-fenotipo en el síndrome de Noonan, si bien ninguno de ellos se presenta de manera uniforme e invariable. La tabla I resume algunas de las asociaciones más aceptadas.

Clínica
El síndrome de Noonan tiene manifestaciones multisistémicas dominadas por la talla baja, la cardiopatía congénita y la facies típica, con alta variabilidad en su expresión.
El síndrome de Noonan se caracteriza por una afectación multisistémica con alta heterogeneidad y expresión clínica variable(8). Existen casos muy leves u oligosintomáticos, que pueden no ser diagnosticados a lo largo de su vida. De herencia autosómica dominante en la abrumadora mayoría de los casos, en los últimos años se ha descrito que algunas variantes de LZTR1 se asocian a herencia recesiva (mientras que otras variantes en este gen dan lugar a fenotipo con la afectación de un solo alelo)(9), y que las variantes en SPRED2 causan una forma autosómica recesiva de síndrome de Noonan(10). El síndrome de Noonan puede considerarse la RASopatía prototípica, y el resto de RASopatías tienen particularidades que permiten distinguirlas. Sin embargo, no existe una manifestación patognomónica de un síndrome, y ninguna manifestación se da en todos los individuos.
El aspecto craneofacial muestra rasgos similares en todas las RASopatías, y es un elemento esencial para la sospecha diagnóstica. Entre otros, destacan: hipertelorismo, ptosis palpebral, oblicuidad palpebral descendente, orejas de implantación baja y antevertidas, cuello ancho y corto con piel redundante en el recién nacido, línea de implantación posterior del cabello baja, facies triangular y diferentes grados de tosquedad facial. Característicamente, el fenotipo facial se va atenuando con la edad(11).
La cardiopatía congénita está presente en cerca de un 80 % de los pacientes, con la estenosis pulmonar valvular y la miocardiopatía hipertrófica como las alteraciones más frecuentes. En tercer lugar están los defectos septales, pero se han descrito todo tipo de cardiopatías, aisladas o combinadas, con un grado de gravedad muy variable(12).
El tamaño al nacimiento es habitualmente normal, con un hipocrecimiento postnatal acusado durante los primeros dos años de vida, en los que es común que se produzca un fallo de medro relacionado con dificultades para la alimentación. Hasta un 20 % de los pacientes pueden precisar sonda nasogástrica o gastrostomía por estos problemas, que tienden a mejorar para el segundo año de vida. El crecimiento posterior se sitúa habitualmente en el percentil 3 con retraso de la edad ósea, y con un estirón puberal tardío y pobre que conduce a una talla por debajo del percentil 3 en un 20-50 % de los adultos(13).
Entre las manifestaciones musculoesqueléticas destacan las anomalías torácicas (pectus excavatum, pectus carinatum, tórax en tonel, teletelia), así como cúbito valgo, genu valgo, escoliosis e hiperextensibilidad articular. Las manifestaciones linfáticas pueden ser más frecuentes de lo que se consideraba clásicamente, y representan la manifestación fetal más frecuente en el síndrome de Noonan, con casos de translucencia nucal aumentada, higroma quístico, quilotórax fetal e hídrops no inmune. Estos y otros hallazgos, como el polihidramnios, a pesar de su baja especificidad, pueden conducir al diagnóstico prenatal de síndrome de Noonan u otras RASopatías(3).
La criptorquidia está presente en cerca de un 60-80 % de los varones afectos, y la fertilidad está disminuida en los varones, mientras que parece estar conservada en las mujeres. Se han descrito también malformaciones renales, como doble sistema colector, riñón único, estenosis pieloureteral y dilatación de la pelvis renal.
El síndrome de Noonan tiene un aumento de riesgo de desarrollar tumores sólidos y síndromes mielodisplásicos que se estima en 8 veces el de la población general(14). Presentan, también, facilidad para hacerse hematomas y una tendencia al sangrado con casos infrecuentes de hemorragia grave. Se han descrito diversos trastornos de coagulación, así como disfunción plaquetaria, aunque a menudo no hay una correlación estrecha entre los resultados de las pruebas de coagulación y la tendencia al sangrado(15).
Mientras que el retraso psicomotor es frecuente, el desarrollo neurocognitivo suele ser favorable, con facultades intelectuales conservadas o solo levemente afectadas. El déficit de atención, las dificultades de aprendizaje y los problemas sociales y emocionales son más comunes que en la población general(16).
Además de las manifestaciones comentadas, el síndrome de Noonan presenta una serie de características adicionales de baja especificidad que ocurren con más frecuencia que en la población general, como estrabismo, defectos de refracción, hipoacusia de transmisión y neurosensorial, manchas café con leche y otras lesiones hiperpigmentadas en la piel, piel seca e hiperqueratosis folicular, y manifestaciones orodentales(3).
Diagnóstico
Aunque el diagnóstico del síndrome de Noonan es clínico, el estudio molecular es una herramienta inestimable en el estudio de pacientes con síndrome de Noonan y otras RASopatías.
A pesar del desarrollo de las técnicas de caracterización molecular, el diagnóstico del síndrome de Noonan sigue siendo clínico, y cerca de un 15-20 % de los pacientes con diagnóstico de síndrome de Noonan no tienen una causa genética identificada. Los criterios más uniformemente aceptados fueron descritos por Van der Burgt en 1994 y revisados en 2007(17) (Tabla II).

De cualquier forma, el estudio molecular se ha convertido en una herramienta indispensable para la orientación diagnóstica de estos pacientes, para la confirmación molecular, como ayuda en el diagnóstico diferencial con el resto de RASopatías, para la guía anticipada basada en los elementos de correlación genotipo-fenotipo, y como ayuda en el estudio familiar(2). En líneas generales se recomienda la realización de un panel de secuenciación masiva que incluya, al menos, los genes implicados en las RASopatías. Menos frecuentemente, o en situaciones de duda, se puede optar por la realización de un exoma o un genoma. La realización de estudios gen a gen mediante secuenciación Sanger no se recomienda, ya que es más costosa y menos eficiente que el panel de genes. Es recomendable que el estudio genético sea orientado por un genetista clínico.
Diagnóstico diferencial
El diagnóstico diferencial incluye las otras RASopatías y otros síndromes no relacionados.
En el diagnóstico diferencial del síndrome de Noonan deben considerarse las otras rasopatías, incluida la neurofibromatosis tipo 1 (Tabla III), así como otros síndromes no relacionados con la vía RAS-MAPK, como el síndrome de Aarskog, el síndrome de Turner o el síndrome de Baraitser-Winter y la familia de las actinopatías.

Tratamiento
El tratamiento en el síndrome de Noonan es fundamentalmente sintomático, ya que no existen tratamientos etiológicos disponibles; sin embargo, el conocimiento de la base molecular de estos trastornos abre la vía para la investigación de tratamientos dirigidos a la raíz de los problemas asociados: la vía de las RAS-MAPKinasas.
En el momento actual, las posibilidades de tratamiento en el síndrome de Noonan y otras RASopatías se limitan al tratamiento sintomático. Desde el año 2020 está aprobado en España, el tratamiento con hormona de crecimiento recombinante humana (rhGH) en el síndrome de Noonan con talla baja (menor de -2,5 DE), por encima de los 2 años de edad. A la hora de iniciar tratamiento con rhGH en el síndrome de Noonan, las deficiencias calóricas deben ser resueltas previamente, debe tenerse en cuenta la patología asociada (seguimiento estrecho de miocardiopatía hipertrófica; vigilancia de escoliosis) y el genotipo (riesgo de neoplasia y de miocardiopatía hipertrófica asociada a algunos genes/mutaciones específicas)(18). Algunos autores recomiendan la realización de una resonancia nuclear magnética cerebral antes de iniciar tratamiento, dado el riesgo de tumores sólidos en estos pacientes.
El conocimiento del sustrato molecular del síndrome de Noonan y otras RASopatías ha abierto el camino para la búsqueda de fármacos etiológicos, dirigidos a atenuar la actividad de la vía RAS-MAPK. Algunos de estos fármacos se emplean ya en cáncer, pero la frecuencia de efectos secundarios y los casos documentados de resistencia dificultan su extrapolación al campo de las rasopatías. Aun así, se han descrito experiencias aisladas favorables con trametinib en pacientes en situaciones clínicas desesperadas, y selumetinib ha sido aprobado por la FDA para el tratamiento de pacientes pediátricos con neurofibromas plexiformes inoperables. El avance en el diagnóstico genético, el desarrollo de modelos animales y el perfeccionamiento de nuevos fármacos, contribuyen al objetivo último de ofrecer alternativas terapéuticas significativas para los pacientes con RASopatías(19).
Prevención
La mayoría de guías de seguimiento publicadas recomiendan un abordaje por distintas áreas de salud, anticipándose a las manifestaciones esperables en función de la edad y el genotipo, cuando existen elementos de correlación genotipo-fenotipo. La tabla IV reúne una síntesis de las recomendaciones de seguimiento aceptadas actualmente(8,17,20). Se recomienda, en general, ofrecer contacto con grupos de apoyo y asociaciones de pacientes y familiares de pacientes con síndrome de Noonan y otras RASopatías, que tienen una actividad importante en el territorio nacional.

Función del pediatra de Atención Primaria
El pediatra de Atención Primaria puede jugar un papel esencial en el diagnóstico, seguimiento y tratamiento de los pacientes con síndrome de Noonan.
El pediatra de Atención Primaria puede ser el primer médico que detecte que un niño tiene síndrome de Noonan. Le pueden hacer sospechar el fallo de medro, la criptorquidia, la talla baja, la detección de un soplo o el antecedente de un trastorno linfoproliferativo. La visión global del paciente por parte del pediatra de Atención Primaria, así como el conocimiento de sus familiares, puede ser clave en la detección temprana. Asimismo, algunas de las evaluaciones que deben hacerse en el seguimiento de un niño con síndrome de Noonan pueden ser solicitadas por el pediatra de Atención Primaria, que puede derivar al paciente a endocrinología pediátrica para valorar tratamiento con rhGH, a urología para tratamiento de criptorquidia, o a Atención Temprana si precisa fisioterapia o terapia ocupacional, entre otras actuaciones.
Conflicto de intereses
No hay conflicto de interés en la elaboración del manuscrito. Declaración de intereses: ninguno.
Bibliografía
Los asteriscos muestran el interés del artículo a juicio del autor.
1.** Tajan M, Paccoud R, Branka S, Edouard T, Yart A. The RASopathy Family: Consequences of Germline Activation of the RAS/MAPK Pathway. Endocr Rev. 2018; 39: 676-700.
2.*** Tartaglia M, Aoki Y, Gelb BD. The molecular genetics of RASopathies: An update on novel disease genes and new disorders. Am J Med Genet C Semin Med Genet. 2022; 190: 425-39.
3.*** Zenker M. Clinical overview on RASopathies. Am J Med Genet C Semin Med Genet. 2022; 190: 414-24.
4. Strullu M, Caye A, Lachenaud J, Cassinat B, Gazal S, Fenneteau O, et al. Juvenile myelomonocytic leukaemia and Noonan syndrome. J Med Genet. 2014; 51: 689-97.
5. Abe Y, Aoki Y, Kuriyama S, Kawame H, Okamoto N, Kurosawa K, et al. Prevalence and clinical features of Costello syndrome and cardio-facio-cutaneous syndrome in Japan: findings from a nationwide epidemiological survey. Am J Med Genet A. 2012; 158A: 1083-94.
6. Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet. 2001; 29: 465-8.
7. Grant AR, Cushman BJ, Cavé H, Dillon MW, Gelb BD, Gripp KW, et al. Assessing the gene-disease association of 19 genes with the RASopathies using the ClinGen gene curation framework. Hum Mutat. 2018; 39: 1485-93.
8.*** Romano AA, Allanson JE, Dahlgren J, Gelb BD, Hall B, Pierpont ME, et al. Noonan syndrome: clinical features, diagnosis, and management guidelines. Pediatrics. 2010; 126: 746-59.
9. Johnston JJ, van der Smagt JJ, Rosenfeld JA, Pagnamenta AT, Alswaid A, Baker EH, et al. Autosomal recessive Noonan syndrome associated with biallelic LZTR1 variants. Genet Med Off J Am Coll Med Genet. 2018; 20: 1175-85.
10. Motta M, Fasano G, Gredy S, Brinkmann J, Bonnard AA, Simsek-Kiper PO, et al. SPRED2 loss-of-function causes a recessive Noonan syndrome-like phenotype. Am J Hum Genet. 2021; 108: 2112-29.
11. Allanson JE. Objective studies of the face of Noonan, Cardio-facio-cutaneous, and Costello syndromes: A comparison of three disorders of the Ras/MAPK signaling pathway. Am J Med Genet A. 2016; 170: 2570-7.
12. Leoni C, Blandino R, Delogu AB, De Rosa G, Onesimo R, Verusio V, et al. Genotype-cardiac phenotype correlations in a large single-center cohort of patients affected by RASopathies: Clinical implications and literature review. Am J Med Genet A. 2022; 188: 431-45.
13.*** Yart A, Edouard T. Noonan syndrome: an update on growth and development. Curr Opin Endocrinol Diabetes Obes. 2018; 25: 67-73.
14.** Villani A, Greer MLC, Kalish JM, Nakagawara A, Nathanson KL, Pajtler KW, et al. Recommendations for Cancer Surveillance in Individuals with RASopathies and Other Rare Genetic Conditions with Increased Cancer Risk. Clin Cancer Res Off J Am Assoc Cancer Res. 2017; 23: e83-90.
15. Di Candia F, Marchetti V, Cirillo F, Di Minno A, Rosano C, Pagano S, et al. RASopathies and hemostatic abnormalities: key role of platelet dysfunction. Orphanet J Rare Dis. 2021; 16: 499.
16. Wingbermühle E, Roelofs RL, Oomens W, Kramer J, Draaisma JMT, Leenders E, et al. Cognitive Phenotype and Psychopathology in Noonan Syndrome Spectrum Disorders through Various Ras/MAPK Pathway Associated Gene Variants. J Clin Med. 2022; 11: 4735.
17.** van der Burgt I. Noonan syndrome. Orphanet J Rare Dis. 2007; 2: 4. Disponible en: https://doi.org/10.1186/1750-1172-2-4.
18.** Stagi S, Ferrari V, Ferrari M, Priolo M, Tartaglia M. Inside the Noonan ‘universe’: Literature review on growth, GH/IGF axis and rhGH treatment: Facts and concerns. Front Endocrinol. 2022; 13: 951331.
19. Saint-Laurent C, Mazeyrie L, Yart A, Edouard T. Novel therapeutic perspectives in Noonan syndrome and RASopathies. 2024; 183: 1011-9.
20.*** Carcavilla A, Suárez-Ortega L, Rodríguez Sánchez A, González-Casado I, Ramón-Krauel M, Labarta JI, et al. Síndrome de Noonan: actualización genética, clínica y de opciones terapéuticas. An Pediatr Barc Spain. 2020; 93: 61.e1-e14.
Bibliografía recomendada
– Tartaglia M, Aoki Y, Gelb BD. The molecular genetics of RASopathies: An update on novel disease genes and new disorders. Am J Med Genet C Semin Med Genet. 2022; 190: 425-39.
Excelente revisión de la historia del descubrimiento de los distintos genes implicados en las RASopatías, de la mano del autor que identificó el primer gen asociado al síndrome de Noonan.
– Zenker M. Clinical overview on RASopathies. Am J Med Genet C Semin Med Genet. 2022; 190: 414-24.
Revisión actualizada de la nosología de las RASopatías, con sus particularidades: breve y claro.
– Yart A, Edouard T. Noonan syndrome: an update on growth and development. Curr Opin Endocrinol Diabetes Obes. 2018; 25: 67-73.
Brillante panorámica de la repercusión del síndrome de Noonan en el crecimiento y desarrollo.
– Noonan syndrome guideline development group. Management of Noonan syndrome. A clinical guideline. Disponible en: https://rasopathiesnet.org/wp-content/uploads/2014/01/265_Noonan_Guidelines.pdf.
Una guía clásica para el seguimiento del síndrome de Noonan, elaborada por Dyscerne, una red de expertos en dismorfología, en 2010 y revisada en 2011. Está en desarrollo una actualización, hasta que llegue esta sigue siendo una ayuda útil.
– Romano AA, Allanson JE, Dahlgren J, Gelb BD, Hall B, Pierpont ME, et al. Noonan syndrome: clinical features, diagnosis, and management guidelines. Pediatrics. 2010; 126: 746-59.
Revisión clásica del síndrome de Noonan con una guía de actuación que, en algunos aspectos se ha quedado un poco antigua, pero ya es un clásico imprescindible.
– Carcavilla A, Suárez-Ortega L, Rodríguez Sánchez A, González-Casado I, Ramón-Krauel M, Labarta JI, et al. Síndrome de Noonan: actualización genética, clínica y de opciones terapéuticas. An Pediatr Barc Spain. 2020; 93: 61.e1-e14.
Actualización de las guías de actuación en síndrome de Noonan, adaptada a nuestro medio.
| Caso clínico |
|
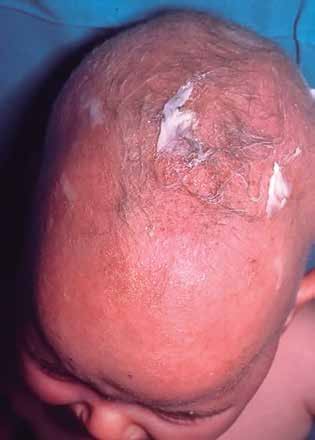
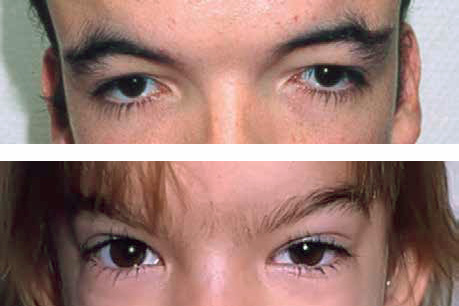
Lactante varón de 7 meses remitido por fenotipo particular y retraso ponderoestatural. Entre sus antecedentes familiares: madre: sin antecedentes de interés; talla: 166 cm. Padre: antecedente de criptorquidia operada; estenosis pulmonar valvular con evolución favorable; talla: 163 cm; diagnosticado en la infancia de síndrome de Dubowitz sin estudio genético. A la exploración física, longitud en -4,83 DE y peso en -3,71 DE. Se aprecia hipertelorismo, ptosis palpebral, raíz nasal hundida, orejas de implantación baja algo grandes e implantación baja del cabello. Presenta también 3 manchas café con leche, pectus excavatum, hígado a 2 cm del reborde costal derecho y no se palpa el testículo izquierdo. No se auscultan soplos. A la exploración, el padre tiene ptosis palpebral e inclinación palpebral descendente. No tiene manchas en la piel ni lunares.
|






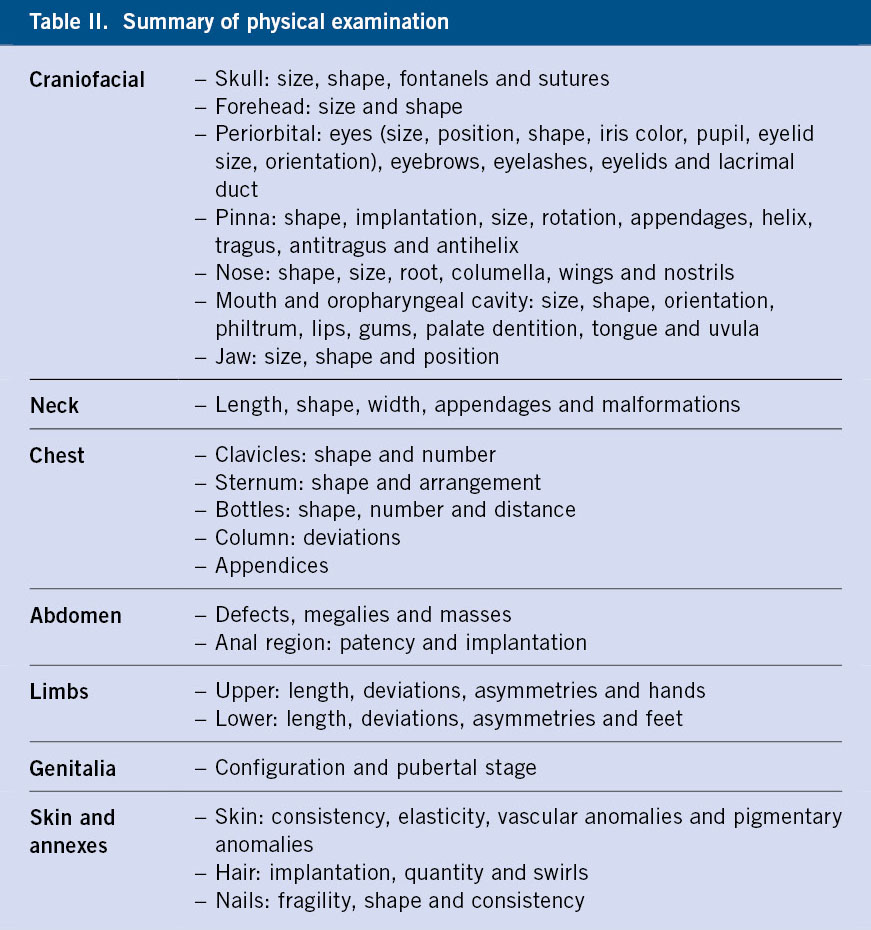
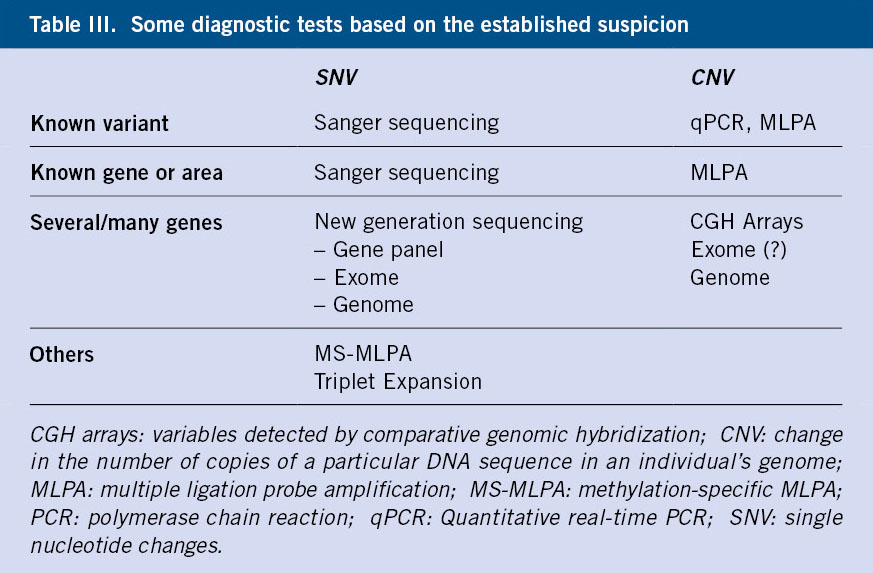










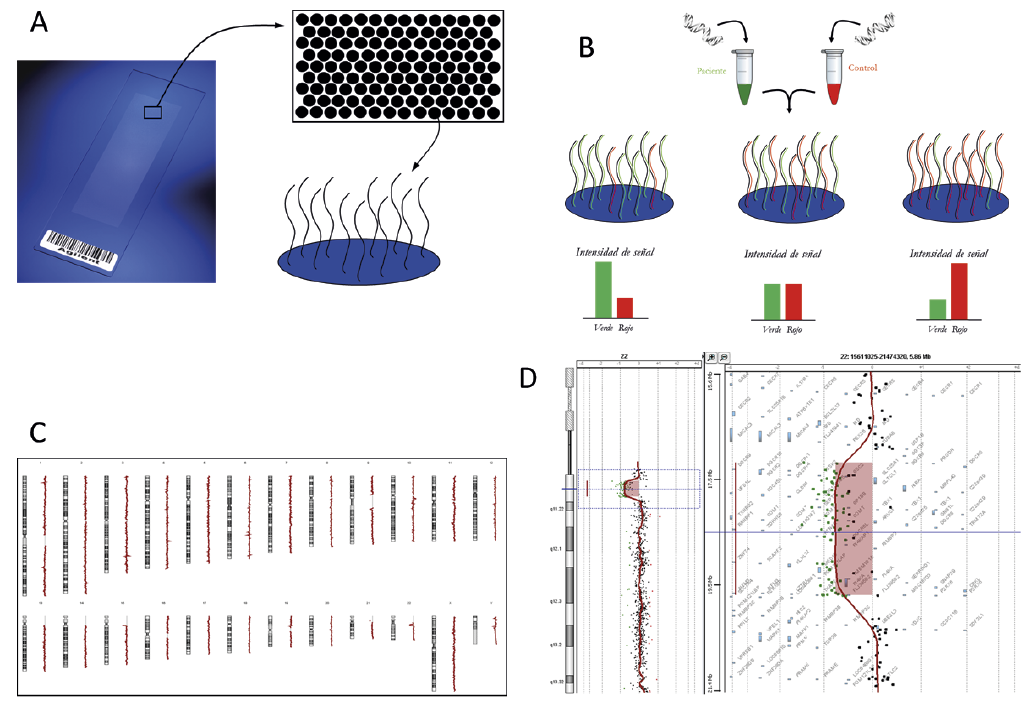

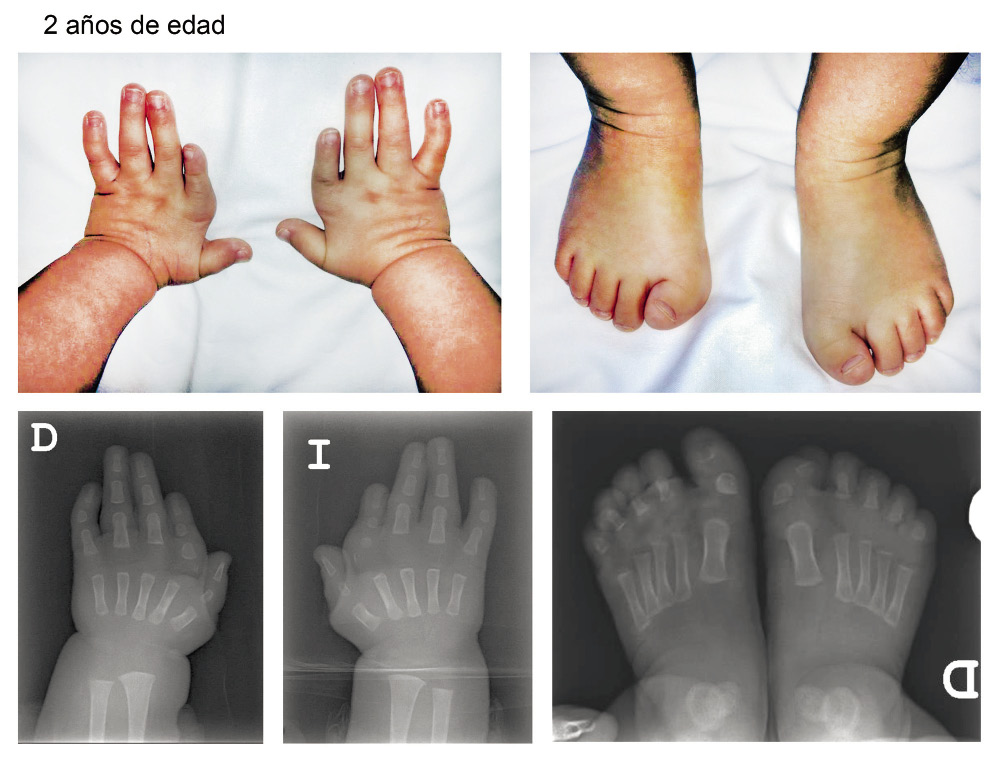














 Figura 1.
Figura 1.
 Figura 2.
Figura 2.