 |
| Temas de FC |
A. Pellicer Martínez, B. Moreno Sanz-Gadea
Servicio de Neonatología, Hospital Universitario La Paz. Madrid
| Resumen
Los vómitos son un signo inespecífico que puede reflejar multitud de enfermedades de diferente gravedad. Aunque las enfermedades banales son las que con mayor frecuencia provocan vómitos, debemos tener especial cuidado con aquellas que pueden ser más graves y comprometer el pronóstico del paciente. Una minuciosa historia clínica, así como una exploración física completa, son fundamentales para orientar el diagnóstico, siendo, en ocasiones, suficientes para su manejo sin necesidad de pruebas complementarias. Es importante también indagar acerca de los antecedentes obstétricos, perinatales y familiares, ya que pueden aportar datos para orientar el diagnóstico. Dependiendo de la presunción diagnóstica, se necesitarán pruebas complementarias, como: analítica sanguínea o pruebas de imagen. |
| Abstract
Vomiting is a non-specific symptom that can reflect a variety of diseases with diverse degrees of severity. Although banal diseases represent the most frequent cause of vomiting, caution should be exerted as more severe conditions might compromise the patient’s prognosis. |
Palabras clave: Vómitos; Estenosis hipertrófica de píloro; Obstrucción intestinal; Hiperplasia suprarrenal congénita; Errores innatos del metabolismo; Alergia PLV.
Key words: Vomiting; Hypertrophic pyloric stenosis; Intestinal obstruction; Congenital adrenal hyperplasia; Inborn errors of metabolism; Cow’s milk protein allergy.
Pediatr Integral 2019; XXIII (3): 138 – 146
Vómitos en el neonato y lactante
Introducción
El vómito es la expulsión del contenido gástrico a través de la boca. Puede aparecer en diversas enfermedades, siendo importante el grupo etario en el que aparecen para orientar de manera correcta el diagnóstico.
Etiología
Los vómitos son un signo inespecífico que puede reflejar multitud de enfermedades.
Los vómitos pueden presentarse en una variedad de enfermedades de gravedad diversa, siendo las más frecuentes aquellas que aparecen en la tabla I(1,2). Aunque las enfermedades banales son las que con mayor frecuencia provocan vómitos, debemos tener especial cuidado con aquellas que pueden ser más graves y comprometer el pronóstico del paciente, como es el caso de las patologías quirúrgicas o las enfermedades metabólicas.

Diagnóstico
En el manejo de los vómitos es imprescindible una correcta historia clínica y una exploración física completa.
Una minuciosa historia clínica, así como una exploración física completa son fundamentales para orientar el diagnóstico del vómito, siendo, en ocasiones, suficientes para su manejo sin necesidad de pruebas complementarias.
Dentro de los datos de la historia clínica, hay que intentar describir sus características: tipo de contenido de los vómitos (gástrico, bilioso, fecaloideo), frecuencia, cantidad, edad de presentación, relación con las tomas, así como síntomas acompañantes como: fiebre, diarrea, alteración neurológica, mal estado general, acidosis metabólica o administración de fármacos. Es importante también indagar acerca de los antecedentes del embarazo y perinatales, así como los antecedentes familiares(1).
Tras una detallada historia clínica, hay que completar con una exploración clínica completa, iniciándola por el estado general del paciente, y continuando por una exploración por aparatos.
Según la orientación diagnóstica, serán necesarias pruebas diagnósticas, como: analítica sanguínea o pruebas de imagen, tipo radiografía abdominal, ecografía, tránsito o enema opaco.
Tratamiento
El tratamiento dependerá de la etiología de los vómitos.
Los fármacos antieméticos no son usados en el periodo neonatal y en el lactante pequeño, quedando relegados a edades más tardías. Lo importante es intentar descubrir la causa de los vómitos para poder tratarlos de manera adecuada. Como medida general, si los vómitos son persistentes, se deberá mantener a dieta absoluta para permitir el reposo del tracto digestivo, iniciando sueroterapia intravenosa para evitar la deshidratación.
Reflujo gastroesofágico
Es fisiológico en periodo neonatal y lactancia temprana. Suele mejorar y/o desaparecer con el crecimiento del bebé y el inicio de la nutrición complementaria.
El reflujo gastroesofágico (RGE) es un trastorno gastrointestinal funcional frecuente, que aparece en los primeros meses de vida, debido a la inmadurez del tracto digestivo, permitiendo el paso del alimento con facilidad desde el estómago hacia el esófago.
Clínica
La clínica va a depender de la edad de aparición y se divide en dos grandes grupos(3):
1. Signos/síntomas digestivos: las regurgitaciones o vómitos son los más frecuentes. También, puede cursar con irritabilidad o rechazo de tomas secundario a esofagitis, que pueden conllevar alteración de la curva ponderoestatural, denominándose enfermedad de reflujo gastroesofágico.
2. Signos/síntomas extradigestivos: en el caso de los recién nacidos y, sobre todo, en los prematuros, es habitual que curse con pausas de apnea seguidas de desaturación y bradicardia coincidente con el momento de las tomas. En los lactantes, en cambio, es más frecuente que asocien infecciones ORL o respiratorias de repetición como: laringitis, otitis, neumonías o broncoespasmos.
Fisiopatología
Aunque se considere un trastorno fisiológico debido a la inmadurez del esfínter esofágico inferior, hay que estar pendientes de la aparición de signos de alarma que sugieran complicaciones del mismo o enfermedades que puedan agravarlo. En el periodo neonatal y en el lactante muy pequeño, especialmente en el prematuro, destacaríamos: los procesos respiratorios, sobre todo, aquellos que precisan de soporte ventilatorio no invasivo, ya que generan un acúmulo de aire en el tracto digestivo que favorece la compresión del diafragma; malformaciones congénitas, como la hernia diafragmática o las atresias intestinales, incluso después de la reparación quirúrgica; o la encefalopatía, cualquiera que sea su origen.
Ciertos fármacos pueden provocar la disminución del tono del esfínter esofágico facilitando el reflujo esofágico. Estos fármacos son usados de manera más o menos frecuente en unidades de Neonatología, como pueden ser: sildenafilo, opioides, teofilina y cafeína.
Diagnóstico
El diagnóstico es clínico, dejando en segundo plano las pruebas complementarias.
El diagnóstico está basado en la clínica, historia clínica y exploración física, siendo suficientes, en la mayoría de los casos, para llegar al diagnóstico. En el periodo neonatal, la realización de pruebas complementarias se reduce a la ecografía abdominal o al tránsito digestivo superior en caso de dudas diagnósticas y para descartar otro tipo de patología del tracto gastrointestinal.
Tratamiento
El pilar fundamental del tratamiento son las medidas posturales y dietéticas.
El tratamiento inicial en cualquier paciente con reflujo gastroesofágico son las medidas posturales, manteniendo una inclinación máxima de 30º del plano del colchón. Aunque la postura en decúbito prono tendría ventajas sobre el supino, únicamente debe limitarse al ámbito hospitalario por su asociación a la muerte súbita del lactante(3).
Tomas más frecuentes, pero menos cuantiosas es otra medida beneficiosa. En aquellos neonatos que precisan ingreso hospitalario y que no tienen autonomía para la alimentación por boca, fundamentalmente prematuros o pacientes con malformaciones mayores, a menudo precisan realizar las tomas de manera más lenta para mejorar el vaciamiento gástrico y disminuir el reflujo. En estos casos, se utilizan bombas de infusión del alimento a través de sonda, en la que se programa la velocidad de forma precisa. La propia sonda nasogástrica mantiene el cardias abierto de manera permanente, por lo que debe ser retirada lo antes posible.
Como se ha comentado anteriormente, el reflujo va mejorando con la edad del bebé debido a la maduración del tracto digestivo, el inicio de la alimentación complementaria y el inicio de la sedestación. La alimentación exclusiva con leche, junto a la posición en decúbito supino del recién nacido, favorecen el paso del alimento desde el estómago al esófago; la diversificación alimenticia, con un alimento más espeso, lo dificulta.
La perpetuación del reflujo importante, refractario a medidas conservadoras, precisa de tratamiento farmacológico(4,5) (Tabla II).

En el neonato, sin embargo, los fármacos utilizados no han demostrado ser eficaces para el tratamiento del RGE, especialmente en el recién nacido prematuro, y pueden asociar efectos secundarios potencialmente graves. A pesar de ello, su uso está muy extendido en las unidades neonatales.
Los principales fármacos usados para el tratamiento del RGE y sus complicaciones en la etapa neonatal son:
• Antisecretores:
- Antagonistas del receptor de histamina H2 (ranitidina): disminuyen la secreción ácida al inhibir los receptores de histamina de las células parietales del estómago.
- Inhibidores de la bomba de protones (omeprazol, esomeprazol, pantoprazol): disminuyen la secreción ácida inhibiendo la bomba Na+/K+ ATPasa de la célula parietal gástrica e inhiben la acción de la gastrina, histamina y agentes muscarínicos.
• Procinéticos (domperidona): aumentan la peristalsis esofágica y aceleran el vaciamiento gástrico.
La cirugía para el RGE más frecuente utilizada es la funduplicatura de Nissen, cuya indicación es la enfermedad grave por RGE, que presenta complicaciones importantes y limita la vida del paciente, o en el caso de pacientes de alto riesgo, como encefalopatías o malformaciones del tracto digestivo superior.
Sepsis
La sepsis es una importante causa de morbimortalidad en los recién nacidos. El cuidado perinatal y el uso juicioso, con indicaciones muy precisas de la antibioterapia materna periparto, han sido determinantes en la reducción de la incidencia de la sepsis precoz de transmisión vertical. Actualmente, el azote de las unidades neonatales es la sepsis nosocomial asociada a la asistencia hospitalaria.
Etiología
Los agentes que con más frecuencia causan sepsis en nuestro medio, se describen en la tabla III.

Clínica
Las manifestaciones clínicas de la sepsis son múltiples y más inespecíficas cuanto menor es la edad del paciente.
Debemos descartar una sepsis ante la presencia de cualquiera de los síntomas siguientes(8,9):
• Respiratorios: pausas de apnea, polipnea, quejido y tiraje.
• Hemodinámicos: taquicardia, bradicardia, hipotensión y mala perfusión.
• Digestivos: rechazo de tomas, vómitos, distensión abdominal y cambio en el patrón de heces.
• Neurológicos: decaimiento, irritabilidad, alteración del nivel de conciencia, hipotonía, ausencia de succión y convulsiones.
• Metabólicas: hipoglucemia, hiperglucemia, acidosis metabólica e ictericia.
• Térmico: hipotermia, hipertermia, gradiente de temperatura central-periférico.
Diagnóstico
La normalidad en las pruebas complementarias no descarta la sepsis.
Ante la sospecha clínica de sepsis, debe actuarse rápidamente para evitar su progresión y posibles consecuencias. El diagnóstico inicial es clínico, aunque nos apoyaremos en los resultados de la analítica de sangre y cultivos(6,9). La prueba de primera línea y la que más información nos puede aportar es la analítica sanguínea, fijándonos especialmente en el recuento leucocitario y plaquetario y en los reactantes de fase aguda.
Sospecharemos sepsis ante(10):
• Leucocitosis (> 20.000 x 10e3/μl) o leucopenia (< 5.000 × 10e3/μl), neutrofilia (> 15.000 × 10e3/μl) o neutropenia (< 1.500 × 10e3/μl) y/o elevación de índice infeccioso ([neutrófilos inmaduros/neutrófilos totales]: > 0,2 si <72 horas; > 0,12 si <72 horas).
• Trombocitopenia (<100.000 x 10e3/μl).
• Proteína C reactiva > 10-20 mg/l.
La procalcitonina es un marcador precoz, ya que se eleva en las primeras 2-4 horas de proceso, y específico. Sin embargo, existe un aumento fisiológico en los primeros días de vida, lo que le resta valor en este periodo. Existen otros marcadores de sepsis, como: las interleucinas (IL-6, IL-8 e IL-10) o el factor de necrosis tumoral alfa (TNF-α), aunque no forman parte de la rutina diagnóstica en la mayoría de unidades neonatales.
El diagnóstico definitivo lo tendremos con la positividad de un cultivo ante un patógeno. Ante una sospecha de sepsis, hemos de tomar muestras para cultivo de los potenciales focos de infección: sangre, orina y líquido cefalorraquídeo. La realización de punción lumbar, en el caso de neonatos con alta sospecha de sepsis con mal estado general, con/sin sintomatología neurológica, y en el caso de bacteriemia confirmada por cultivo, es obligatoria, ya que la meningitis puede darse en estos pacientes, sin clínica neurológica específica o muy sutil.
En el caso de pacientes hospitalizados portadores de tubo endotraqueal y catéter central, hay que descartar que el origen de la sepsis no sea secundario a una bronconeumonía asociada a ventilación mecánica o a sepsis por catéter. En estos casos, además, cultivaremos el aspirado broncoalveolar y la punta del catéter una vez retirado, respectivamente. En la búsqueda del origen de la infección y dependiendo del contexto, se toman, así mismo, muestras de la placenta, región recto-vaginal o leche materna.
La sepsis en los primeros meses de vida suele ser secundario a patología bacteriana, aunque no de forma exclusiva, ya que diferentes virus pueden dar un cuadro clínico similar a la sepsis(11,12). Su estudio también debe ser realizado cuando se sospecha que el origen de la sepsis puede ser vírico. Esto es especialmente aplicable a las infecciones por virus respiratorios en los prematuros(11,12).
Tratamiento
El inicio de tratamiento antibiótico no debe demorarse por la realización de pruebas complementarias.
Según el inicio de la clínica y la edad del paciente, se iniciarán antibióticos dirigidos a cubrir los gérmenes más frecuentes en esa etapa. Una vez obtengamos los cultivos y si alguno de ellos resulta positivo, se modificará el tratamiento para dirigirlo al germen concreto. La duración del mismo va a depender del origen y tipo de microorganismo implicado.
Alergia a proteínas de la leche de vaca
La alergia a proteínas de leche de vaca (APLV) es una reacción de hipersensibilidad mediada por mecanismo inmunológico. Es una enfermedad que está aumentando de manera importante en los últimos años en todo el mundo, siendo su prevalencia actual de un 0,5-3%(13,14). Suele aparecer en los primeros meses de vida y desaparecer en la mayoría de los pacientes a los 5 años de edad. Presentan reacciones cruzadas con proteínas de leche de otros mamíferos.
Los alérgenos principales en este tipo de alergia son: la caseína y las proteínas séricas, como α-lactoalbúmina, β-lactoglobulina y seroalbúmina.
Clínica
La anafilaxia es la forma más grave de APLV.
Los síntomas acompañantes van a depender de si se trata de reacciones inmediatas, que aparecen desde los pocos minutos hasta las dos horas de la ingesta del alérgeno, con mayor riesgo vital; o reacciones tardías, más allá de las dos horas, incluso a los días de la toma de leche.
Entre los síntomas que pueden presentarse se encuentran(13,14):
• Cutáneos: angioedema, urticaria y dermatitis.
• Respiratorios: broncoespasmo y estridor.
• Digestivos: regurgitaciones, vómitos, diarrea, dolor abdominal, prurito oral, heces con sangre y estancamiento ponderal.
• Neurológicos: alteración del nivel de conciencia y síncope.
• Hemodinámicos: hipotensión y shock.
Diagnóstico
La mejoría clínica tras la dieta de exclusión apoya el diagnóstico de APLV.
El diagnóstico se establece a través de la historia clínica del paciente, sensibilización a proteínas de leche de vaca a través de prick-test, determinación en suero de IgE específica y mejoría clínica tras la supresión de la leche.
Tratamiento
La eliminación de las proteínas de leche de vaca en la dieta es el tratamiento principal.
La dieta exenta en proteína de leche de vaca es la primera medida a realizar ante la sospecha de alergia; en muchos casos, incluso antes del diagnóstico de certeza.
En el caso de los bebés alimentados con lactancia materna exclusiva, se explicará a las madres que deben excluir esa proteína de su dieta (leche y derivados) para poder mantener la lactancia. Aquellos alimentados con leche artificial, deberán cambiar la fórmula por una extensamente hidrolizada. Se reservará la leche elemental para los niños de alto riesgo o que no toleren la anterior.
Estenosis hipertrófica de píloro
La estenosis hipertrófica de píloro es la obstrucción al vaciamiento gástrico a nivel pilórico secundaria a hiperplasia e hipertrofia de las capas musculares de esa zona. Es una patología típica del lactante pequeño, apareciendo de manera más frecuente alrededor de la tercera-quinta semana de vida. Se estima una prevalencia de 1-5 casos/1.000 lactantes, siendo más frecuente en varones, primogénitos y raza blanca(3).
Etiopatogenia
Se desconoce la causa exacta; si bien, se presume un origen multifactorial, con una predisposición genética y exposición a factores ambientales.
Entre los factores implicados, se encuentran el tabaquismo materno durante el embarazo o la alimentación con biberón. Se ha descrito una mayor frecuencia entre gemelos o hermanos, con tendencia a la agregación familiar. Se ha asociado al consumo de macrólidos en los últimos meses del embarazo, al paso de los mismos a través de la leche materna o a su administración en menores de 2 semanas(3,15,16).
Clínica
El cuadro clínico típico es un neonato varón de 3-4 semanas de vida que presenta vómitos posprandiales no presentes previamente.
Los vómitos son de carácter propulsivo y de contenido alimenticio, repetidos y se presentan tras las tomas. El momento del comienzo de esta sintomatología es importante para orientar el diagnóstico. Suelen mantener el apetito, cogiendo con avidez las tomas. Si los vómitos persisten, pueden aparecer signos y síntomas de deshidratación.
Diagnóstico
La prueba diagnóstica definitiva es la ecografía abdominal.
En ocasiones, en la palpación abdominal, en el cuadrante superior derecho, puede apreciarse la “oliva pilórica”, masa dura, lisa y ovalada que refleja el engrosamiento a nivel del píloro. Ante la sospecha, la prueba principal a realizar es la ecografía abdominal, en la que se visualiza con objetividad un grosor del píloro mayor de 4 mm, una longitud mayor de 15 mm y el estrechamiento del canal (Fig. 1)(3,16).
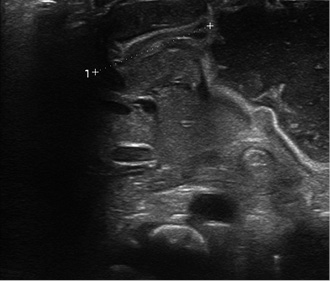 |
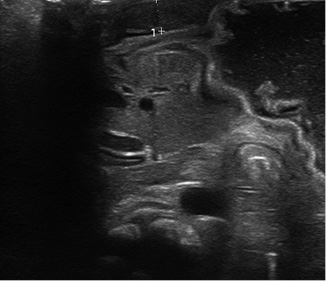 |
Figura 1. Aumento del grosor y longitud del píloro.
En la analítica sanguínea, se objetiva alcalosis metabólica hipoclorémica secundaria a la pérdida de ácido clorhídrico a través de los vómitos(3,16). Según el grado de deshidratación asociada, se podrá ver alterada la función renal o el hematocrito.
Tratamiento
El tratamiento quirúrgico debe de realizarse tras la normalización de las alteraciones hidroelectrolíticas.
La cirugía no es urgente, teniendo que corregirse previamente las alteraciones hidroelectrolíticas. La técnica quirúrgica es la pilorotomía extramucosa de Ramstedt(3,16), mediante abordaje laparoscópico umbilical o supraumbilical. El pronóstico es muy bueno y la recuperación suele ser rápida.
Obstrucción intestinal
La aparición de vómitos en la primera semana de vida, siempre tiene que hacernos pensar en una anomalía congénita del tracto digestivo que cause obstrucción intestinal. Existen múltiples causas de obstrucción intestinal en el neonato y lactante pequeño(3), y todas ellas constituyen una urgencia quirúrgica, ya que supone un compromiso agudo de la vascularización regional. Entre las causas posibles, cabe destacar: la atresia intestinal, la enfermedad de Hirschsprung y la malrotación intestinal +/- vólvulo intestinal.
Clínica
La presencia de vómitos biliosos debe orientar el diagnóstico siempre hacia la obstrucción intestinal.
La clínica principal de obstrucción intestinal son los vómitos que, si bien son más específicos los de contenido bilioso, podrían ser de contenido alimenticio inicialmente. Pueden acompañarse de distensión abdominal. El paso de meconio no excluye estos diagnósticos. Tanto el vómito como las heces, pueden contener cantidades variables de sangre, según la etiología(3,17). La etiología del cuadro obstructivo y el tiempo de evolución determinarán el grado de deterioro del estado general, que puede llegar a ser crítico, con inestabilidad hemodinámica y deterioro neurológico. Constituye, por tanto, una urgencia vital en algunos casos, sobre todo, cuando hay compromiso de la circulación esplácnica, como es el caso del vólvulo.
Diagnóstico
La radiografía y ecografía abdominal son las pruebas iniciales ante la sospecha de una obstrucción intestinal.
Ante una clínica compatible con obstrucción intestinal, la radiografía de abdomen es la primera prueba a realizar (Fig. 2).

Figura 2. Signo de doble burbuja.
Los hallazgos iniciales pueden ser inespecíficos o sutiles, dando la apariencia de normalidad, por lo que la ecografía abdominal es obligadamente complementaria. Es útil para localizar los vasos mesentéricos y descartar malrotación y posible vólvulo. La normalidad de las pruebas de imagen no descarta obstrucción. La vigilancia clínica es obligada. La intervención quirúrgica es la única prueba confirmatoria.
La analítica de sangre es una herramienta de apoyo al diagnóstico de isquemia intestinal. Inicialmente, es inespecífica y, a medida que evoluciona la enfermedad, aparece elevación de reactantes de fase aguda y, secundariamente a la destrucción celular, puede haber aumento de amilasa, LDH, CPK y transaminasas. También, puede aparecer acidosis metabólica con elevación de ácido láctico y alteraciones electrolíticas(17).
En el caso de la enfermedad de Hirschsprung, se produce obstrucción debido a la ausencia congénita de neuronas parasimpáticas de la pared intestinal del recto-sigma o incluso de segmentos más amplios del colon o intestino delgado. La expresión clínica, por tanto, es muy variable y va desde el retraso en la evacuación de meconio a un cuadro de obstrucción intestinal grave. El diagnóstico incluye el enema opaco y la confirmación precisa de la realización de biopsia intestinal. La manometría no es concluyente en la etapa neonatal.
Tratamiento
La obstrucción intestinal supone una urgencia quirúrgica.
El tratamiento inicial es mantener dieta absoluta, colocación de sonda nasogástrica gruesa abierta a bolsa, para favorecer la evacuación del contenido gástrico, y el inicio de sueroterapia. La cirugía se realiza de manera urgente y, según los hallazgos encontrados, se realizarán o no resecciones, tipo de anastomosis u ostomía de descarga. Todos estos pacientes son subsidiarios de alimentación parenteral de duración variable, dependiendo de las causas de la obstrucción, hasta conseguir una autonomía para la alimentación enteral.
Hiperplasia suprarrenal congénita
La hiperplasia suprarrenal congénita es una enfermedad de herencia autosómica recesiva que causa un fallo en la esteroidogénesis suprarrenal.
Etiopatogenia
El déficit de 21-hidroxilasa es la forma más frecuente de hiperplasia suprarrenal congénita.
Hay diferentes formas que varían en gravedad, siendo el déficit de cortisol el punto común a todas ellas. La consecuencia es un aumento en la hormona ACTH con la consiguiente estimulación de la corteza suprarrenal, generando un aumento de esteroides previo al bloqueo enzimático.
Las formas descritas son: forma clásica con pérdida salina, forma clásica sin pérdida salina y forma no clásica. En este capítulo, nos centraremos en la forma clásica con pérdida salina, ya que es el tipo que debuta en el periodo neonatal presentando, entre otros, un cuadro digestivo, siendo, además, la forma de hiperplasia suprarrenal congénita más grave. Dentro de la forma clásica, el déficit de 21-hidroxilasa supone el 95% de los casos. El defecto de 21-hidroxilasa hace que la esteroidogénesis suprarrenal se desvíe del bloqueo para generar andrógenos, con una disminución en la síntesis de mineralcorticoides y glucocorticoides(18).
Clínica
El cuadro característico es un recién nacido con pérdida salina y alteración de la diferenciación sexual o macrogenitosomía, según se trate de hembra o varón, respectivamente.
La enfermedad debuta en los primeros días de vida con un cuadro clínico de: vómitos, disminución del apetito, escasa ganancia ponderal y poliuria. Pueden presentar taquicardia e hipotensión, así como signos de deshidratación.
El hiperandrogenismo secundario al bloqueo enzimático produce en las niñas virilización de genitales externos en grado variable y en los niños macrogenitosomía(18-20).
Es un cuadro grave, que puede poner en riesgo la vida del paciente si no se instaura el tratamiento correcto.
Diagnóstico
El cribado metabólico del recién nacido de algunas comunidades autónomas incluye el déficit de 21-hidroxilasa.
Ante un recién nacido con alteraciones en la diferenciación sexual y alteración neurológica, debemos pensar en esta entidad. Presenta alteraciones analíticas, con: acidosis metabólica, hipoglucemia, hiponatremia, hiperpotasemia, natriuresis elevada, disminución de aldosterona, elevada actividad de renina plasmática y cociente elevado de actividad renina plasmática/aldosterona.
Ante la sospecha de déficit de 21-hidroxilasa, se deben medir los niveles de 17-hidroxiprogesterona que se encuentran muy elevados, variando el rango según la edad(18-20).
En el estudio genético, se analizará el gen responsable del déficit de 21-hidroxilasa, denominado CYP21A2, que se localiza en el complejo mayor de histocompatibilidad HLA, en el brazo corto del cromosoma 6 (6p21.3)(20).
Tratamiento
El tratamiento primordial es la administración de glucocorticoides y mineralcorticoides.
Además del tratamiento sintomático asociado a las complicaciones agudas, desde el primer momento, hay que iniciar el tratamiento sustitutivo con corticoides, ya que es su déficit el que pone en riesgo vital al paciente. El glucocorticoide más utilizado es la hidrocortisona, de baja potencia y vida media corta, superponible al cortisol endógeno. La dosis para hiperplasia suprarrenal congénita es 15 mg/m2/día, administrada cada 8 horas vía oral, precisando los neonatos dosis mayores (50 mg/m2/día). La 9-fludrocortisona es el mineralcorticoide de elección en estos casos. La dosis en neonatos y lactantes es 0,1-0,2 mg/kg, repartida, por vía oral, cada 12 horas. Ante situaciones de estrés leve, se debe duplicar-triplicar la dosis de glucocorticoide hasta 24 horas después del cese del proceso(18). Además, hasta el inicio de la alimentación complementaria se añaden suplementos de cloruro sódico oral (2-4 mEq/kg/día) divididos en varias tomas.
La otra parte del tratamiento es la cirugía correctora de los genitales. Aunque el tiempo en el que se debe realizar no está consensuado, se recomienda realizarlo antes de los 15-18 meses de vida.
Enfermedades congénitas del metabolismo
Las enfermedades congénitas del metabolismo son un conjunto de enfermedades producidas por una alteración genética que causa defectos estructurales o funcionales en la síntesis de una enzima o en su cofactor. Estos defectos pueden conllevar déficit o acumulación de metabolitos, causantes de la sintomatología. Son trastornos poco frecuentes de manera individual, pero su incidencia aumenta cuando se consideran colectivamente todos ellos.
Clínica
Las manifestaciones clínicas son muy variables, por lo que es importante mantener un alto índice de sospecha.
Los signos y síntomas de enfermedades metabólicas son muy diversos, variando la forma de presentación según la edad y tipo de metabolopatía.
El debut en época neonatal implica mayor gravedad y suele estar precedido por un intervalo libre de síntomas, que puede ir desde horas hasta días, siendo la presentación aguda una urgencia metabólica. Entre las alteraciones clínicas que pueden aparecer, encontramos las siguientes(21-23):
• Neurológicas: succión débil, hipotonía, debilidad muscular, letargia, irritabilidad, coma, convulsiones y apnea.
• Metabólicas: acidosis metabólica, hiperlactatemia, cetosis y deshidratación.
• Hepáticas: hepatomegalia, hiperbilirrubinemia, hipoglucemia, hiperamoniemia, hipertransaminasemia, colestasis y fallo hepático.
• Gastrointestinales: vómitos y rechazo de tomas.
• Cardiacas: cardiomiopatía y arritmias.
• Oculares: cataratas y opacidad corneal.
• Otros: rasgos dismórficos, hidrops fetalis y olor anormal.
Diagnóstico
Una primera muestra de sangre con estudio básico puede orientar hacia una enfermedad metabólica.
Parte de los pacientes son diagnosticados a través del cribado metabólico del recién nacido; pero en otros, la sintomatología debuta sin una orientación previa. Información acerca de los antecedentes familiares (muerte por causa desconocida), consanguinidad familiar y antecedentes obstétricos (hiperémesis gravídica, disfunción hepática y síndrome HELLP), pueden ayudar a su diagnóstico.
Un primer análisis básico de sangre puede ser suficiente para apoyar nuestra sospecha diagnóstica, teniendo que extraer posteriormente diferentes muestras para poder clasificar la enfermedad(21,22):
• Sangre:
- Básico: hemograma, coagulación, ionograma, calcio, gasometría, glucosa, perfil hepático, ácido úrico, colesterol, amonio y cuerpos cetónicos.
- Metabólico: lactato, piruvato, 3OH-butirato, acetoacetato, ácidos grasos no esterificados, aminoácidos, carnitina, acilcarnitinas, transferrina deficiente en hidratos de carbono, homocisteína, ácidos grasos de cadena muy larga y esteroles.
• Orina:
- Básico: glucosa, pH, cuerpos cetónicos, electrolitos, ácido úrico, creatinina, sustancias reductoras y test de sulfitos.
- Metabólico: aminoácidos, ácidos orgánicos, ácido orótico, guanidinoacético, purinas, pirimidinas, mucopolisacáridos, oligosacáridos, sales biliares, polioles y sulfátidos.
Se deberá hacer ampliación de los estudios según la orientación diagnóstica, incluyendo otros líquidos biológicos, como: líquido cefalorraquídeo y biopsias. En algunas ocasiones, hay que recurrir a las muestras de tejidos y estudios post mortem.
Tratamiento
El tratamiento específico suele instaurarse tras la confirmación diagnóstica del tipo de metabolopatía.
El tratamiento inicial consiste en la suspensión de la alimentación enteral y aporte de nutrientes vía parenteral, con una restricción proteica. En los casos en los que el debut ha sido agudo y con riesgo vital, generalmente se precisa: soporte respiratorio, hemodinámico, corrección de alteraciones hidroelectrolíticas o metabólicas, o eliminación de metabolitos acumulados mediante procedimientos específicos de depuración extrarrenal. La terapia específica se inicia tras la confirmación diagnóstica; si bien, al tener algunas enfermedades metabólicas formas vitamino-dependientes, algunos cofactores seleccionados se pueden administrar previamente al diagnóstico de confirmación como: biotina, tiamina, vitamina B12, riboflavina y piridoxina(21).
Síndrome de abstinencia
El síndrome de abstinencia se define como el conjunto de signos y síntomas sistémicos secundarios a la interrupción brusca de la exposición a determinados fármacos o drogas. Puede deberse a la exposición de dicha sustancia intraútero, tras consumo materno, o estar asociado a la retirada de tratamientos intrahospitalarios en niños con procesos complejos que precisan sedoanalgesia.
El consumo materno de este tipo de sustancias puede asociarse a otros indicadores de morbilidad fetal, como: parto prematuro, crecimiento intrauterino retardado, malformaciones congénitas, procesos disruptivos del sistema nervioso central, alteraciones del neurodesarrollo o muerte(24).
Clínica
Los rasgos dominantes del síndrome son las alteraciones digestivas, de la neuroconducta y neurovegetativas.
El inicio, duración y gravedad van a depender del tipo de sustancia, dosis y tiempo de exposición. Existen escalas que valoran la gravedad del síndrome de abstinencia y que ayudan en el manejo del mismo (Tabla IV-V)(24-26).
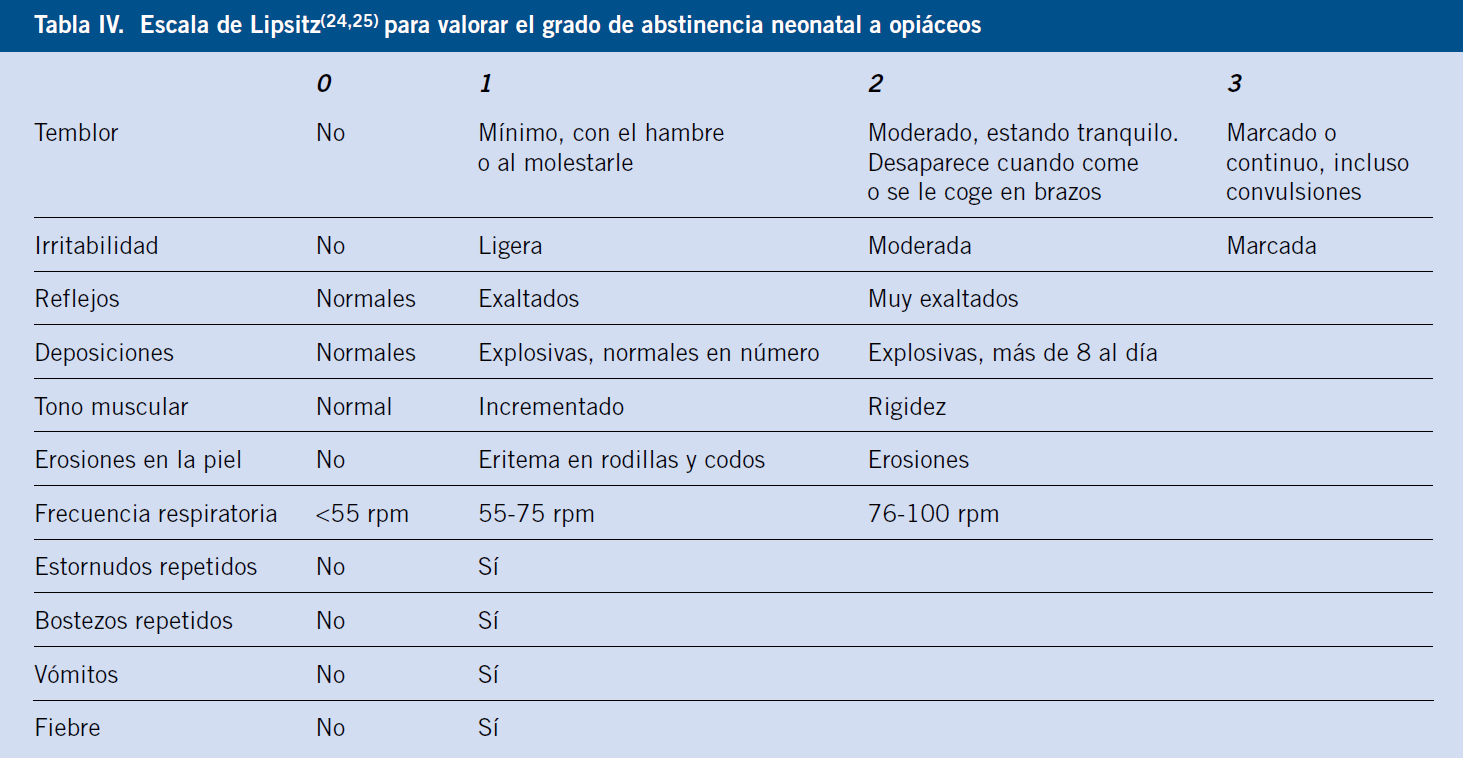
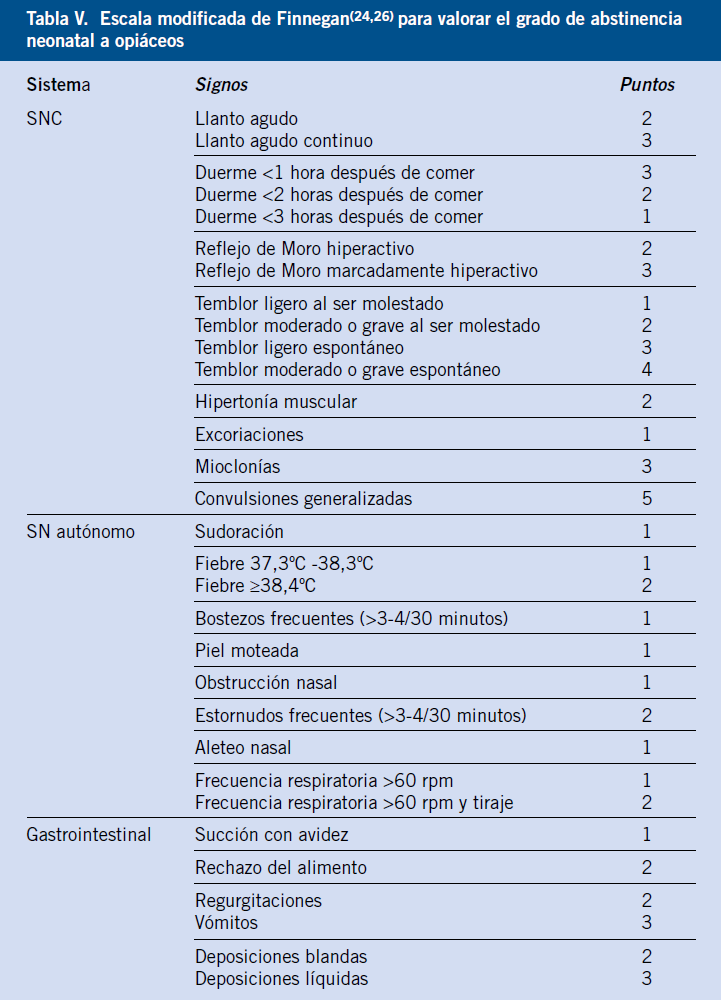
Se considera síndrome de abstinencia significativo, la puntuación de escala de Lipsitz >4 o escala de Finnegan >8. Esto permite adecuar el tratamiento.
Diagnóstico
El diagnóstico es exclusivamente clínico.
En el caso de exposición intraútero a sustancias o sospecha de consumo materno no declarado, se debe recoger muestra de orina para estudio de tóxicos.
Tratamiento
Se debe favorecer un ambiente tranquilo y evitar la estimulación excesiva en pacientes con síndrome de abstinencia.
El tratamiento inicial deben ser medidas no farmacológicas: mantener ambiente tranquilo, medidas de contención, favorecer la lactancia materna siempre y cuando no se contraindique por consumo materno de determinadas sustancias y método canguro. Si estas medidas no son suficientes, se utilizarán medidas farmacológicas(4,5,24) (Tabla VI).

El descenso de la medicación prescrita se hará acorde a la evolución de la puntuación de las escalas.
Bibliografía
Los asteriscos reflejan el interés del artículo a juicio del autor.
1. Di Lorenzo C. Approach to the infant or child with nausea and vomiting. UpToDate. Disponible en: http://www.uptodate.com.
2. Raske ME, Dempsey ME, Dillman JR, Dory CE, Garber M, Hayes LL, et al. ACR Appropriateness Criteria Vomiting in Infants up to 3 Months of Age. J Am Coll Radiol. 2015; 12(9): 915-22.
3. Vázquez Fernández ME, Cano Pazos M. Vómitos y regurgitaciones, reflujo gastroesofágico y estenosis pilórica. Pediatr Integral. 2015; XIX (1): 21-32.
4.*** Young TE, Margum B. Neofax. Manual de drogas neonatológicas. Panamericana.
5.*** Pediamecum AEP. Disponible en: pediamecum.es.
6. Morven S Edwards, MD. Clinical features, evaluation, and diagnosis of sepsis in term and late preterm infants. UpToDate. Disponible en: http://www.uptodate.com.
7.** De la Torre M, De Lucas N, Velasco R, Gómez B, Mintegi S, Grupo para el estudio del lactante febril de la Red de investigación de la Sociedad Española de Urgencias de Pediatría. Etiología y evolución de las infecciones potencialmente graves en lactantes menores de 3 meses febriles. An Pediatr (Barc). 2017; 87(1): 42-9.
8.** Goldstein B, Giroir B, Randolph A. International pediatric sepsis consensus conference: Definitions for sepsis and organ dysfunction in pediatrics. Pediatr Crit Care Med. 2005; 6: 2-8.
9. Sánchez García L, Elorza Fernández D. Recién nacido con riesgo infeccioso. Actitud diagnóstica. An Pediatr Contin. 2011; 9(4): 239-48.
10.*** Hofer N, Zacharias E, Müller W, Resch B. An Update on the Use of C-Reactive Protein in Early-Onset Neonatal Sepsis: Current Insights and New Tasks. Neonatology. 2012; 102: 25-36.
11. Kidszun A, Hansmann A, Winter J, Gröndahl B, Knuf M, Weise K, Mildenberger E. Detection of respiratory viral infections in neonates treated for suspicion of nosocomial bacterial sepsis: a feasibility study. Pediatr Infect Dis J. 2014; 33(1): 102-4.
12. Pichler K, Assadian O, Berger A. Viral Respiratory Infections in the Neonatal Intensive Care Unit-A Review. Front Microbiol. 2018; 9: 2484.
13. Lapeña López de Armentia S, Naranjo Vivas D. Alergia a proteínas de leche de vaca. Pediatr Integral. 2013; XVII(8): 554-63.
14.** Koletzko S, Niggemann B, Arato A, Dias JA, Heuschkel R, Husby S, et al. Diagnostic Approach and Management of Cow’s-Milk Protein Allergy in Infants and Children. J Pediatr Gastroenterol Nutr. 2012; 55: 221-9.
15.** Zhu J, Zhu T, Lin Z, Qu Y, Mu D. Perinatal risk factors for infantile hypertrophic pyloric stenosis: A meta-analysis. J Pediatr Surg. 2017; 52(9): 1389-97.
16. Olivé AP, Endom EE. Infantile hypertrophic pyloric stenosis. UptoDate. Disponible en: http://www.uptodate.com.
17. Moreno Sanz-Gadea B, Udaondo Gascón C, Sellers Carrera M, Martín Sánchez J, De Ceano-Vivas La Calle M. Lactante con vómitos, ¿cuándo sospechar un vólvulo intestinal? An Pediatr (Barc) 2018; 88: 109-11.
18. Labarta Aizpún JI, De Arriba Muñoz A, Ferrández Longás A. Hiperplasia suprarrenal congénita. Protoc diagn ter pediatr. 2011; 1: 117-28.
19.** Gutiérrez Pascual L, Guerrero-Fernández J. Insuficiencia suprarrenal. Crisis adrenal. En: Manual de diagnóstico y terapéutica en Pediatría. Panamericana. 2017. p. 861-9.
20. Rodríguez Sánchez A, Sanz Fernández M, Echeverría Fernández M. Hiperplasia suprarrenal congénita por déficit de 21-hidroxilasa. Pediatr Integral. 2015; XIX (7): 488-97.
21.*** Ellaway CJ, Wilcken B, Christodoulou J. Clinical approach to inborn errors of metabolism presenting in the newborn period. J Paediatr Child Health. 2002; 38: 511.
22. Pedrón Giner C, Rubio Cabezas O. Signos sugestivos de metabolopatía congénita. En: Urgencias y tratamiento del niño grave. Ergón; p. 1412-6.
23.** Palacios A, García O, García-Silva MT. Diagnóstico de los errores innatos del metabolismo. An Pediatr Contin. 2008; 6: 347-52.
24. Marín Huarte N, Blanco Sánchez AI, Sánchez Salmador R, Gómez Martín F. Recién nacido de madre con patología no infecciosa. En: Manual de diagnóstico y terapéutica en Pediatría. Panamericana. 2017. p. 1806-13.
25. Lipsitz PJ. A proposed narcotic withdrawal score for use with newborn infants. A pragmatic evaluation of its efficacy. Clin Pediatr (Phila). 1975; 14(6): 592-4.
26. Finnegan LP, Connaughton JF Jr, Kron RE, Emich JP. Neonatal abstinence syndrome: assessment and management. Addict Dis. 1975; 2(1-2): 141-58.
27.*** Kocherlakota P. Neonatal abstinence syndrome. Pediatrics. 2014; 134(2): e547-61.
Bibliografía recomendada
- Young TE, Margum B. Neofax. Manual de drogas neonatológicas. Panamericana.
Manual muy útil de consulta de fármacos en neonatos.
- Kocherlakota P. Neonatal abstinence syndrome. Pediatrics. 2014; 134(2): e547-61.
Artículo muy interesante acerca del síndrome de abstinencia en neonatos. Describen el inicio y duración del síndrome de abstinencia secundario a diversas sustancias/fármacos, así como su manejo farmacológico con sus posibles efectos secundarios.
| Caso clínico |
|
Neonato de 19 días de vida que acude a urgencias por vómitos alimenticios desde hace 24 horas. Los padres refieren que, desde los primeros días de vida, ha presentado regurgitaciones frecuentes, pero que desde hace 1 día han empeorado, apareciendo en la mayoría de las tomas, aunque no impresiona de rechazo de tomas. Realiza 3 deposiciones al día, de consistencia normal. No presenta fiebre ni otra clínica acompañante. Tiene una hermana de 2 años con infección respiratoria de vías altas. Antecedentes perinatales Embarazo controlado. Serología materna: inmune a rubeola, resto negativas. Ecografías prenatales normales. Parto vaginal, eutócico, Apgar 9/10, pH en arteria umbilical: 7,29. Piel-con-piel inmediato postparto. Lactancia materna exclusiva. Exploración física Buen estado general sin signos de deshidratación. Buen color y perfusión. Auscultación cardiopulmonar normal. Abdomen: blando, no doloroso, no megalias. Neurológico: vital, reactivo. Tono y actitud acordes a edad. Reflejos primitivos presentes. Fontanela anterior normotensa. Pruebas complementarias Gasometría: pH: 7,37; pC02: 50,5 mmHg; bicarbonato: 29,2 mmol/L; exceso de bases: 2,9 mmol/L; Na: 137 mmol/L; K: 5,1 mmol/L; Cl: 99 mmol/L. Evolución Dado el buen estado general, se mantiene en observación. Realiza una toma presentando nuevamente vómito alimenticio cuantioso, por lo que se deja a dieta absoluta y se inicia sueroterapia intravenosa.
|
