|
|
|
|
|


D. González-Lamuño Leguina*, M.L. Couce Pico**
*Servicio de Pediatría, Hospital Universitario Marqués de Valdecilla-Universidad de Cantabria. Santander. **Unidad de Diagnóstico y Tratamiento de Enfermedades Metabólicas Congénitas, Servicio de Neonatología, Hospital Clínico Universitario de Santiago, Departamento de Pediatría, IDIS, CIBERER, Santiago de Compostela, La Coruña, España
| Resumen
El cribado durante el período neonatal incluye determinados procesos de detección pre-sintomática de enfermedades o trastornos que, sin una sintomatología aparente, pudieran causar graves problemas físicos, mentales o del desarrollo, y en los que un diagnóstico y tratamiento precoces, mejoran significativamente su pronóstico. En la actualidad, en el cribado neonatal, se realiza una única determinación sanguínea a las 48 horas de vida. En España, está recomendado realizar el cribado neonatal en muestra de sangre impregnada en papel para, al menos, 8 entidades: hipotiroidismo congénito (HC), fenilcetonuria (PKU), deficiencia de acil CoA deshidrogenasa de ácidos grasos de cadena media (MACDD), deficiencia de 3-hidroxi acil CoA deshidrogenasa de ácidos grasos de cadena larga (LCHADD), aciduria glutárica tipo I (AG-1), deficiencia de biotinidasa (BTD), fibrosis quística (FQ) y anemia de células falciformes (AF). La incorporación de la espectrometría de masas en tándem (MS/MS) posibilita ampliar los trastornos a cribar. También está indicado realizar cribado de hipoacusia congénita. Tanto la recogida de la muestra de la gota de sangre en papel como el cribado de hipoacusia, se realizan antes del alta en la Maternidad. En determinadas situaciones de riesgo (prematuridad, gemelaridad y niños graves), debe hacerse una segunda determinación para disminuir, en lo máximo posible, el número de falsos negativos. |
| Abstract
Screening during the neonatal period includes certain processes of pre-symptomatic detection of diseases or disorders that, without an apparent symptomatology, could cause serious physical, mental or developmental problems, and in which an early diagnosis and treatment significantly improves their prognosis. Currently, a single blood determination is made at 48 hours of age for the neonatal screening. In Spain, the recommendation to perform the neonatal screening involves a blood sample impregnated on paper for at least 8 entities: congenital hypothyroidism (CH), phenylketonuria (PKU), Medium-chain acyl-CoA dehydrogenase deficiency (MCAD), Long chain 3-hydroxyacyl-CoA dehydrogenase deficiency (LCHADD), glutaric aciduria type I (GA-1), biotinidase deficiency (BD), cystic fibrosis (CF) and sickle cell anemia. The incorporation of Tandem mass spectrometry (MS / MS), makes it possible to expand the disorders to be screened for. Congenital hypoacusis screening is also indicated. Both, the drop of blood on paper collection and the screening for hearing loss, are performed before hospital discharge. In certain risk situations (prematurity, twins and severely ill children), a second determination must be made to reduce, as much as possible, the number of false negatives |
Pediatr Integral 2019; XXIII (3): 169.e1 – 169.e10
Cribado neonatal
Introducción
El cribado durante el período neonatal, hace referencia a determinados procesos para detectar en el recién nacido (RN) enfermedades o trastornos que, sin una sintomatología aparente, pudieran causar graves problemas físicos, mentales o del desarrollo, y en los que un diagnóstico y tratamiento precoces, mejoran significativamente su pronóstico.
Aunque existen diferentes programas de cribado neonatal (PCN) diseñados en función de la organización asistencial y recursos destinados a los programas de prevención en cada Comunidad, todos ellos están reconocidos en los diferentes sistemas sanitarios, como programas esenciales de prevención en Salud Pública. Estos programas están integrados en la actividad asistencial de las diferentes unidades pediátricas, por lo que los PCN no deben identificarse solo con un procedimiento técnico o de laboratorio, sino con una actividad coordinada del sistema sanitario que asegure su eficacia y eficiencia.
Son sinónimos de cribado los términos: tamizaje, despistaje, detección precoz o screening, que la Organización Mundial de la Salud (OMS) define como: “la identificación presuntiva, con la ayuda de unas pruebas, de exámenes, o de otras técnicas susceptibles de aplicación rápida, de los sujetos afectados por una enfermedad o por una anomalía que hasta entonces había pasado desapercibida”.
En la actualidad, en España, está recomendado realizar el cribado neonatal en muestra de sangre impregnada en papel para, al menos, 8 entidades: hipotiroidismo congénito (HC), fenilcetonuria (PKU), deficiencia de acil CoA deshidrogenasa de ácidos grasos de cadena media (MACDD), deficiencia de 3-hidroxi acil CoA deshidrogenasa de ácidos grasos de cadena larga (LCHADD), aciduria glutárica tipo I (AG-1), deficiencia de biotinidasa (BTD), fibrosis quística (FQ) y anemia de células falciformes (AF). También está indicado realizar cribado de hipoacusia congénita (Tabla I).
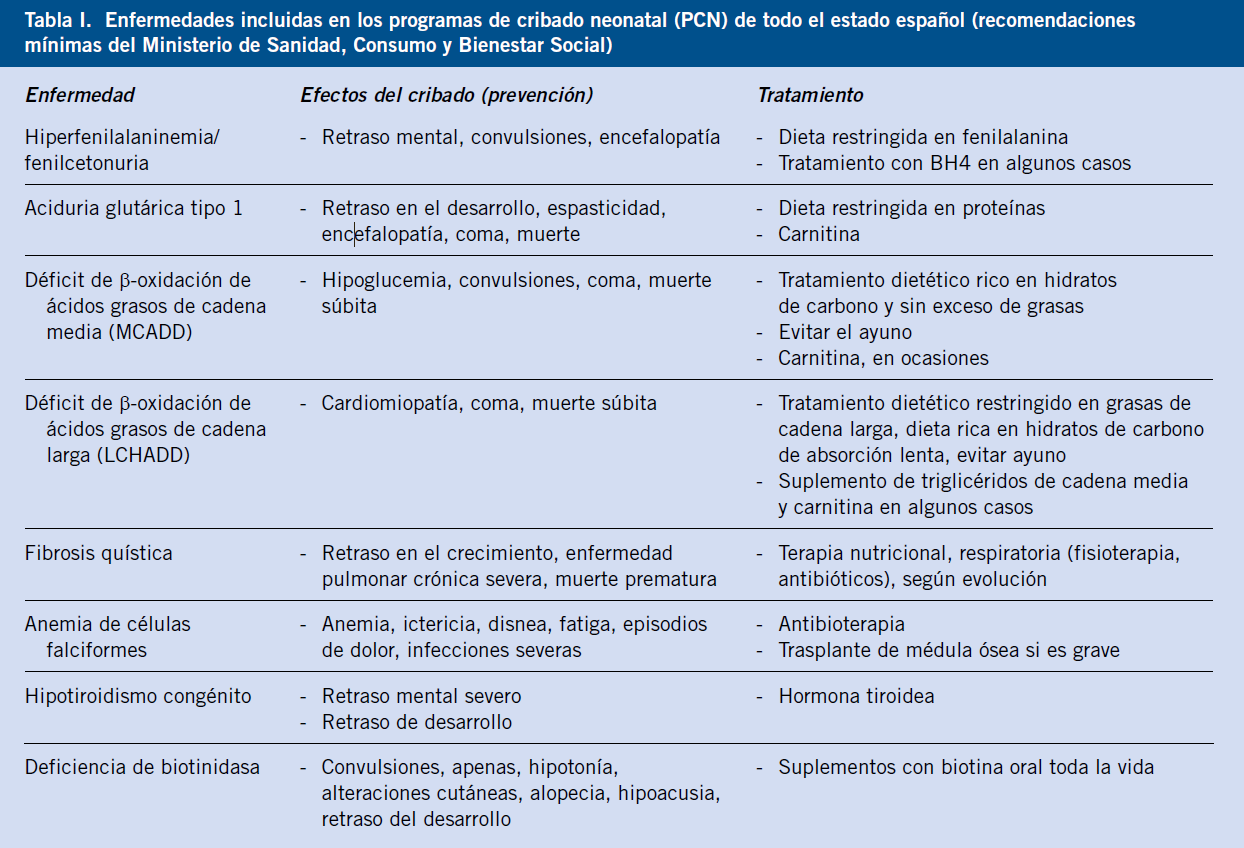
Además, de acuerdo a la evidencia actual, existe la recomendación de incluir la enfermedad de la orina con olor a jarabe de arce (MSUD), acidemia isovalérica (IVA) y homocistinuria (HCY)(1).
Los inicios del cribado neonatal se remontan a la década de los años 60, con el desarrollo de una prueba bacteriológica capaz de detectar en una gota de sangre seca, recogida en papel de filtro, una elevación de los niveles de fenilalanina (test de Guthrie)(2). En España, los diferentes PCN se inician en la década de los años 70, y en 1978, desde el Ministerio de Sanidad, se formaliza a nivel estatal el Programa de Detección Precoz Neonatal de Fenilcetonuria e Hipotiroidismo Congénito.
El momento de realización de las PCN ha ido variando de acuerdo al tipo de análisis y técnica disponible, coexistiendo hasta hace pocos años PCN con muestra única de sangre seca, tomada al 2º o 3er día de vida, y otros con muestra doble (a las 24 horas y a partir del 5º día de vida). Señalar que el término de cribado de enfermedades endocrino-metabólicas, en la actualidad, es inadecuado, ya que a los PCN se ha incorporado el despistaje de la fibrosis quística, hemoglobinopatías, en algunos centros inmunodeficiencias, y la detección precoz de la hipoacusia, rebasando, por tanto, el término de “pruebas del talón” popularmente acuñado.
La incorporación en los laboratorios de cribado de técnicas analíticas basadas en la espectrometría de masas en tándem (MS/MS), ha condicionado cambios significativos en el número de enfermedades potencialmente despistadas, lo que ha dado lugar al denominado cribado metabólico expandido. La MS/MS es una tecnología que detecta y cuantifica de forma simultánea más de 50 metabolitos presentes en la muestra de gota de sangre seca, lo que posibilita el cribado simultáneo de más de 40 errores congénitos del metabolismo (ECM)(3).
El propósito de los análisis utilizados en el cribado neonatal es identificar a todos los RN presuntamente positivos y clasificarlos respecto a la probabilidad de que tengan un trastorno concreto, con un mínimo aceptable de resultados falsos positivos. Esto implica establecer un punto de corte arbitrario para los analitos/marcadores cuantificados. Establecer un punto de corte muy elevado significa perder de forma inaceptable una proporción del grupo diana, mientras que establecer uno demasiado bajo significa exponer a demasiadas personas a la investigación, la mayoría no afectas.
En este punto, señalar que los PCN no son procedimientos de diagnóstico definitivo, identifican grupos de personas de alto riesgo a las que se les ofrece un diagnóstico. Los individuos que presenten un resultado positivo requerirán procedimientos diagnósticos posteriores y, para ello, los PCN deben integrarse con unidades clínicas y de laboratorio especializadas en el diagnóstico y el tratamiento de cada una de las enfermedades sometidas a cribado. En general, el objetivo de los PCN es establecer el diagnóstico correcto e inicio de tratamiento de los trastornos objeto de cribado en los primeros 10 días de vida.
Requisitos para el cribado de una enfermedad
El beneficio principal de un PCN es la prevención de discapacidades asociadas a la enfermedad. Por ello, se recomienda realizar el cribado neonatal de las enfermedades en las que se haya demostrado claramente el beneficio de la detección temprana.
Los PCN tienen diferentes sistemas organizativos basados en su adaptación a las distintas estructuras sanitarias, con la finalidad de obtener la máxima calidad analítica y cobertura. La estrategia se deberá planificar, de forma que se alcance una cobertura del 100% de los RN y el tratamiento temprano del 100% de los casos detectados. Clásicamente, se definieron unos criterios para que un trastorno se incluya en el screening masivo: que la gravedad de la enfermedad curse con morbilidad (retraso mental) o mortalidad si no se diagnostica en el período neonatal; que exista un tratamiento eficaz; que la frecuencia de la enfermedad sea relativamente elevada (al menos, 1 de cada 10.000-15.000 RN); y que exista un método analítico de cribado rápido, fiable y de bajo coste (Wilson & Jungner 1968)(4). Estos criterios serían garantía para que se cumpla el objetivo principal de este tipo de programas: “el máximo beneficio con el mínimo de costes”.
Aunque de forma global estos criterios permanecen vigentes, tras la incorporación de la tecnología de MS/MS, la prevalencia de la enfermedad evaluada sería la suma de la prevalencia de cada una de las enfermedades detectadas con la misma prueba(5). Bajo esta premisa, podría considerarse eficiente incluir el cribado para enfermedades con muy baja prevalencia (< 1:50.000 RN) detectadas mediante MS/MS(6). De acuerdo a estas y otras consideraciones, en el año 2005, la Academia Americana de Pediatría (AAP) y la Asociación Americana de Genética Médica (American College of Medical Genetics, ACMG) emitieron una recomendación para los EE.UU. con los trastornos que deberían despistarse en los PCN. Se incluye un panel principal con 29 patologías genéticas tratables y, en un segundo nivel, 25 patologías más, que también serían susceptibles de ser incluidas en los PCN (Tabla II)(7,8).

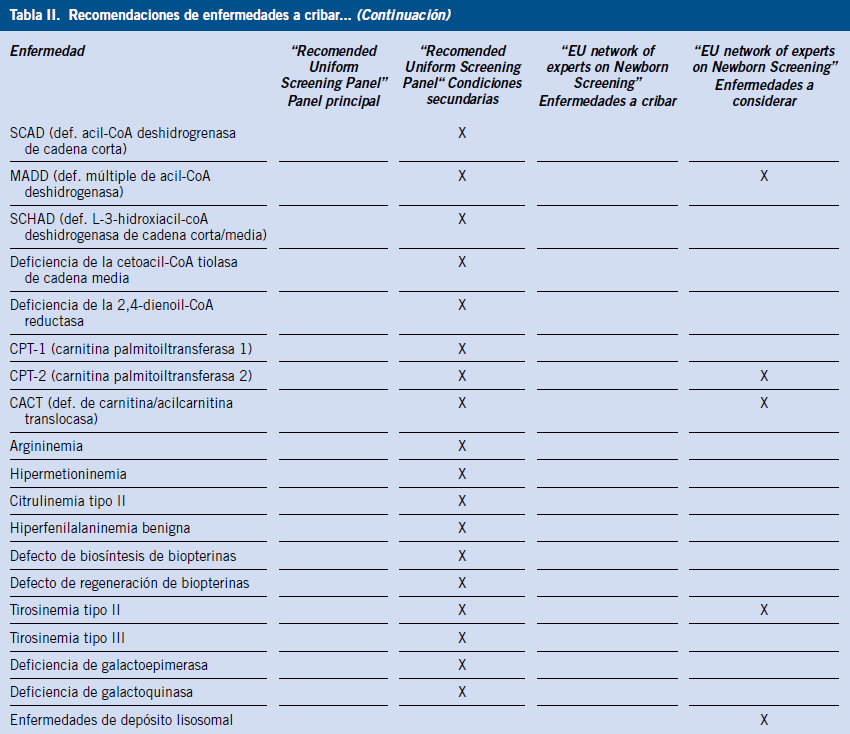
De acuerdo a estas recomendaciones, en algunas regiones, se está cribando para más de 50 condiciones genéticas, a pesar de que, en muchas ocasiones, ni el pediatra ni otros profesionales de Atención Primaria están suficientemente familiarizados con dichas condiciones. Es predecible que, en un futuro próximo, los programas de screening adoptarán diferentes tecnologías de cribado que ampliarán, aún más, el número de trastornos congénitos cribados(9,10).
Obtención de la muestra de sangre para el cribado neonatal
La obtención de muestras de sangre sobre papel absorbente está estandarizada y requiere seguir unas determinadas pautas, utilizando el material adecuado y específico para evitar, en la medida de lo posible, la obtención de muestras no válidas(11).
Aunque las estrategias de obtención de las muestras biológicas puedan diferir, como norma general, se recomienda una extracción única de sangre capilar a partir de las 48 h de vida del RN, tras una ingesta de 24 horas de alimento. Con el fin de garantizar una cobertura del 100% de los RN y gracias a la optimización de los puntos de corte y ratios entre diferentes analitos, la tendencia general es la de realizar la toma de muestra antes del alta hospitalaria del RN.
La extracción de sangre capilar la deberá realizar exclusivamente personal sanitario y nunca los padres, siendo la punción del talón el procedimiento habitual. Para minimizar el dolor del procedimiento de punción, se recomienda administrar durante el procedimiento: una toma de lactancia materna, soluciones de glucosa o chupetes impregnados en sacarosa. Otras intervenciones no farmacológicas que pueden ayudar a disminuir el dolor son: favorecer el contacto visual con los padres, la estimulación táctil, el contacto piel con piel o la succión no nutritiva.
La punción debe realizarse con lanceta estéril con punta < 2,4 mm en la porción medial o lateral de la superficie plantar del talón, para evitar daños osteo-articulares; debe haberse masajeado el pie previamente para aumentar el flujo sanguíneo en la zona y desinfectado con alcohol de 70º, evitando el uso de derivados iodados. La venopunción en el dorso de la mano es un procedimiento alternativo en lactantes y niños, que permite obtener un mayor volumen y con un menor riesgo de que la muestra se hemolice o coagule.
Después de la punción, se debe limpiar la primera gota de sangre con una gasa estéril, dejar que se forme una nueva gota grande de sangre y que esta caiga sobre el papel absorbente de forma que la sangre se absorba y llene el círculo por completo con una sola aplicación (Fig. 1).

Figura 1. Modo de recogida de muestra de sangre en papel.
Debe aplicarse la sangre solamente en uno de los lados del papel, examinando ambos lados para asegurarse de que la sangre haya traspasado uniformemente el papel. Cada laboratorio deberá especificar el número de círculos de sangre que deben ser rellenados.
No es recomendable el procedimiento de recoger la gota con un capilar y, posteriormente, dejarla caer sobre el papel, ya que este método aumenta el número de muestras sobresaturadas, así como el riesgo de rascar y levantar parte de la fibra del papel con el capilar.
La sangre se recoge en un papel de filtro absorbente denominado, genéricamente, tarjeta de Guthrie, que cumpla con los estándares establecidos (Muntkell 2014/07, S&S y Whatman 903™, Ahlstrom). Para que la muestra sea óptima, las manchas de sangre que se obtienen deben contener, al menos, 75 μl (13 mm de diámetro, aproximadamente) y deben dejarse secar en una superficie horizontal no absorbente, seca y limpia durante, al menos, una hora a temperatura ambiente, evitando la luz solar directa. Las muestras, una vez obtenidas, deben enviarse al laboratorio lo antes posible, a poder ser dentro de las 24-48 h siguientes a la extracción. Se debe evitar, siempre que sea posible, el contacto entre las muestras, utilizando, si es necesario, un papel separador entre las tarjetas. Debe evitarse tocar o manchar las gotas de sangre con agua, desinfectantes, jabones o alcohol, para evitar cualquier tipo de contaminación e interferencias. Todos los aspectos mencionados repercuten negativamente en la calidad del proceso.
También es posible realizar estudios de cribado a partir de muestras de orina en papel de filtro, utilizando técnicas analíticas basadas en MS/MS o en cromatografía de gases y espectrometría de masas (GC/MS), que permiten: la confirmación diagnóstica, complementar la información de la muestra de sangre o ampliar horizontes a nuevos diagnósticos. En algunas regiones de Canadá y países asiáticos, se han iniciado PCN en muestras de orina. En España, los PCN incorporan muestras de orina en 3 comunidades autónomas.
Necesidad de segundas muestras de sangre
Aunque, en la mayoría de las ocasiones, se realiza una única determinación, hay situaciones de riesgo en que se debe hacer una segunda toma de muestra, con el fin de disminuir en lo máximo posible el número de falsos negativos.
• Prematuros/bajo peso y gemelos monocigóticos: se recomienda una segunda toma de muestra de sangre entre la 2ª y 4ª semana de vida en los prematuros de menos de 34 semanas y/o 1.500 g de peso y gemelos monocigóticos.
• RN con patología grave al nacimiento: se recomienda tomar una muestra en el momento del ingreso (si es posible antes de que puedan recibir tratamiento antibiótico o alimentación parenteral) y repetirla a las 48-72 horas de vida, según resultados. En estas situaciones de estrés, pueden existir elevaciones transitorias de TSH, tripsina inmunorreactiva o 17-hidroxiprogesterona.
Merecen además especial atención, otras situaciones clínicas, como son las transfusiones previas a la toma de muestra cribado, que obligan a realizar una toma de muestra a las 48-72 horas de la transfusión y repetirla a los 120 días. En los RN alimentados con alimentación parenteral, debe realizarse una toma de muestra a las 48-72 horas después de haber iniciado la alimentación enteral, con independencia de la edad del niño en ese momento. Por último, en niños que hayan precisado exploraciones con contrastes yodados o sometidos a cirugía, debe repetirse la muestra a las 48-72 horas.
Detección del hipotiroidismo congénito (HC)
El hipotiroidismo congénito (HC) es el resultado de una disminución de la actividad biológica tisular de las hormonas tiroideas, bien por producción deficiente, ya sea a nivel hipotálamo-hipofisario (hipotiroidismo central) o a nivel tiroideo (hipotiroidismo primario), o bien por resistencia a su acción o alteración de su transporte en los tejidos diana (hipotiroidismo periférico).
El HC tiene una importancia extraordinaria en el niño por su potencial repercusión sobre su desarrollo intelectual, dado que las hormonas tiroideas son imprescindibles para el desarrollo cerebral, siendo esta la causa de retraso mental prevenible más frecuente. La frecuencia de la enfermedad en 1/3.500 RN vivos, justifica la existencia de un programa de cribado neonatal. La enfermedad que es diagnosticada precozmente con inicio de tratamiento dentro del primer mes de vida se ha asociado a coeficientes de inteligencia dentro de los límites de la normalidad, sin presentar problemas de aprendizaje, y con crecimiento satisfactorio. La detección del HC no debe ser solo precoz, sino urgente, y la administración inicial de L-tiroxina no debe ser únicamente a dosis sustitutivas, sino a dosis terapéuticas.
Existen formas de HC transitorio, debidas a prematuridad, utilización de compuestos yodados o consumo materno de ciertos medicamentos. En este tipo de hipotiroidismo, la función tiroidea se normaliza en un tiempo variable.
El cribado sistemático neonatal se basa en la determinación del nivel de TSH en sangre obtenida del talón de los RN y depositada en cartulinas de papel de filtro. El nivel de TSH, por definición, está siempre elevado (≥ 10µUI/ml) en el hipotiroidismo primario. El estudio de confirmación de los casos positivos o dudosos se realiza mediante la medida de los niveles séricos de T4 libre, que habitualmente está descendido, pero que, en algunos casos, puede ser normal. La ecografía y gammagrafía tiroideas, medida de la concentración sérica de tiroglobulina, determinación del título de anticuerpos antitiroideos y yoduria, esclarecen la etiología.
Existen situaciones de alteración hormonal intermedia, con valores elevados de TSH con T4 normal (hipertirotropinemia con TSH≥10 µUI/ml y tT4>6 µg/dl), o valores de TSH normal con T4 baja (hipotiroxinemia con TSH<10 µUI/ml y tT4<6 µg/dl). Estos casos intermedios, de carácter habitualmente transitorio, pueden precisar tratamiento durante un tiempo, por lo que son enviados directamente, sin precisar segunda muestra, al Servicio de Endocrinología pediátrica para valoración y diagnóstico.
Aún con resultados normales, hay algunas situaciones especiales en las que será necesario repetir las determinaciones a los 15 días de vida: prematuros <33 semanas de gestación y/o peso <1.500 g y en los gemelos univitelinos(12).
Cribado metabólico expandido
La tecnología de MS/MS automatizada es un procedimiento por el cual se pueden identificar simultáneamente una gran cantidad de enfermedades metabólicas (más de 40 ECM) en una misma muestra, muchos de las cuales son adecuadamente tratables. El análisis de determinados metabolitos (acil-carnitinas y aminoácidos) en la sangre desecada y, más aún, la posibilidad de estudiar el perfil de aminoácidos y ácidos orgánicos en la orina del recién nacido, permite detectar mediante MS/MS varios trastornos del metabolismo de los aminoácidos, ácidos orgánicos y ácidos grasos, de forma simultánea.
El paradigma de los EIM incluidos en los PCN es la PKU, una enfermedad del metabolismo de la fenilalanina (Phe) que se transmite de forma autosómica recesiva. Su prevalencia oscila entre 1/12.000-1/18.000 RN, según se valoren las formas de PKU clásica o las formas moderadas/hiperfenilalaninemias (HFA). El cribado se realiza midiendo por MS/MS la Phe y tirosina (Tyr) en sangre extraída a las 48-72 horas de vida del RN, impregnada en papel absorbente. Esta tecnología, al tener una alta sensibilidad, ha permitido adelantar la toma de muestras respecto a la utilizada antiguamente, donde era necesario que el niño o niña hubiera ingerido alimentación, al menos, 4-5 días. El punto de corte es Phe ≥ 2,5 mg/dl o 151,5 µmol/L.
Los trastornos de la beta oxidación mitocondrial son EIM de los ácidos grasos, bien por mutaciones en MCADD o en LCHADD, que se trasmiten de forma autosómica recesiva. La prevalencia de estos dos trastornos es muy variable, entre 1/9.000 y 1/100.000 RN. Como consecuencia de estos fallos, se origina un acúmulo de ácidos grasos, así como de sus derivados conjugados, acilcarnitinas y acilglicinas en sangre y orina. Esto dará lugar a graves problemas fisiopatológicos, a un defecto de energía y/o a un acúmulo de moléculas tóxicas. La clínica aparece en situaciones de descompensación metabólica, cuando hay un aumento de las necesidades energéticas o disminuye el aporte alimenticio. Estas enfermedades son consideradas catastróficas si no son detectadas en el período neonatal, ya que el ayuno prolongado ocasiona la muerte en un 25 a 30% de estos pacientes, y el tratamiento consiste en mantener una alimentación frecuente, evitando el ayuno prolongado.
La AG-1 es una enfermedad del metabolismo de los ácidos orgánicos por defecto de la enzima GCDH (glutaril-CoA-deshidrogenasa), que se transmite con herencia autosómica recesiva, con una prevalencia en España de 1/85.000 RN. Esta enzima es la encargada de metabolizar los aminoácidos lisina (Lys) y triptófano (Trp), productos del metabolismo de las proteínas. Si esta enzima falta o es defectuosa, se produce un bloqueo en la reacción, acumulándose compuestos muy tóxicos para el sistema nervioso. Al nacimiento, el bebé no presenta problema, pero a medida que inicia la alimentación, algunos aminoácidos no se degradan bien y los derivados tóxicos comienzan a acumularse. Un proceso infeccioso, fiebre o ayuno prolongado suelen desencadenar la enfermedad, manifestándose como una crisis encefalopática (convulsiones, irritabilidad, hipotonía…). El cribado disminuye la morbimortalidad, al permitir establecer un diagnóstico precoz, y permite la posibilidad de realizar consejo genético y prevenir nuevos casos en la familia.
Por último, recientemente se ha incorporado el BTD, una forma de aparición tardía del déficit múltiple de carboxilasas, un error congénito del metabolismo que, si no se trata, se caracteriza por convulsiones, dificultad para respirar, hipotonía, erupciones en la piel, alopecia, pérdida de audición y retraso en el desarrollo. La prevalencia del déficit de biotinidasa (BTD) clínico se estima en 1/61.000. La frecuencia de portadores en la población general es aproximadamente de 1/120. Los síntomas suelen aparecer en los primeros meses de vida, pero también se ha descrito una aparición posterior. Los individuos con un déficit profundo sin tratar (menos del 10% de la actividad promedio normal de biotinidasa en suero) presentan hallazgos clínicos variables incluyendo: convulsiones, hipotonía, erupciones eccematoides, alopecia, ataxia, pérdida de audición, infecciones por hongos y retraso en el desarrollo. Metabólicamente, los niños sin tratar pueden mostrar acidosis cetoláctica, acidemia (aciduria) orgánica e hiperamonemia leve. Los individuos con déficit de BTD parcial sin tratar (10-30% de la actividad promedio normal de BTD) pueden ser asintomáticos, pero durante periodos de estrés, como enfermedad, fiebre o ayuno, pueden desarrollar síntomas similares a los de los individuos con un déficit de BTD profundo. El tratamiento primario consiste en suplementos con biotina oral en forma libre, no unida a una proteína: mejora los síntomas en pacientes sintomáticos y previene los síntomas en pacientes identificados mediante el cribado neonatal o antes de que los síntomas se desarrollen. Una vez que se han desarrollado algunos rasgos como atrofia óptica, pérdida de audición, o retraso en el desarrollo, puede que ya no sean reversibles con el tratamiento con biotina. El tratamiento con biotina debe mantenerse de por vida. No se conocen efectos adversos serios de la terapia con biotina. El pronóstico para los individuos diagnosticados es muy bueno, siempre que sean tratados antes de la aparición de los síntomas y cumplan con la terapia de biotina.
Estos programas de cribado metabólico ampliado han obligado a un cambio tecnológico de las Unidades de Bioquímica y suponen un acercamiento o traslación de conocimientos o aplicaciones básicas y especialidades a la práctica asistencial habitual(13).
Actitud ante un cribado positivo de metabolopatía
1. Resultados alterados, altamente sugestivos de una alteración metabólica grave: se debe contactar lo antes posible con la familia, mediante llamada telefónica por parte de un profesional con experiencia en el manejo de enfermedades metabólicas. Idealmente, se trataría de un clínico que conozca la evolución, pronóstico y tratamientos disponibles para dichas enfermedades, que pueda explicar a los padres el significado del resultado y los pasos a seguir para confirmar o excluir el diagnóstico. Pudiera ser necesario actuar de forma urgente, utilizando medidas de depuración o fármacos huérfanos que deben ser aplicados en Unidades con experiencia en el manejo de los errores innatos del metabolismo.
2. Resultados fuera del rango normal, pero no tan alterados como para indicar una enfermedad: estos casos sospechosos requieren una repetición del test para distinguir entre el que tiene una enfermedad (verdadero positivo) del que habitualmente tiene una anomalía transitoria motivada por la inmadurez de algunos sistemas y órganos del RN. Generalmente, la solicitud de nueva muestra se realiza a través de una carta que envía el laboratorio de cribado, en la que se explica que el resultado inicial es anormal. Puede sugerirse la posibilidad de un trastorno y que es necesaria una muestra adicional para descartar un resultado positivo verdadero. En todo caso, en esta carta, se aconseja a los padres que acudan a su pediatra, responsable de asegurar la repetición del test y de proporcionar una explicación adicional a los padres, así como de informar a estos si deben adoptar alguna medida especial con su bebé (p. ej.: evitar ayunos prolongados)(14).
Cribado de fibrosis quística
La fibrosis quística ha sido considerada candidata para ser cribada en el RN, además del lógico cribado genético en cascada de posibles portadores a partir del caso índice.
El argumento para el cribado de RN se basa en que un tratamiento temprano mejora el pronóstico; por lo que, actualmente, se encuentra dentro del PCN universal recomendado (Tabla I). El método está basado en la medida en sangre en papel de la tripsina inmunorreactiva (TIR), la cual está incrementada en la fibrosis quística.
La tripsina se encuentra elevada en edades tempranas de la enfermedad, debido a la obstrucción de los conductos pancreáticos exocrinos y estas cifras se mantienen altas al cabo de los 28 días de vida en los pacientes afectos de la enfermedad. La determinación se realiza en sangre seca, mediante técnicas de: radioinmunoensayo (RIA), inmunofluorescencia a tiempo retardado (DELFIA) o enzimoinmunoensayo (ELISA).
Los resultados falsos positivos de esta determinación pueden ser debidos a la raza o el estado de portador. En cuanto a los falsos negativos, se ven influenciados por la edad de realización de la prueba y por la presencia de íleo meconial.
Como método de screening, es un método sensible, pero con insuficiente especificidad, por lo que se ha adoptado de forma más generalizada, un protocolo en dos etapas para reducir el número de falsos positivos y mejorar el valor predictivo positivo. No existe consenso acerca de la estrategia a utilizar y, como consecuencia, se observa una elevada variabilidad entre los diferentes PCN.
Entre las estrategias de cribado llevadas a cabo, se encuentran: una determinación inicial de TIR seguida de análisis de ADN para las mutaciones más frecuentes o una determinación inicial de TIR seguida de una segunda determinación de TIR a los 20-30 días si la primera ha sido elevada y posterior análisis de ADN solo a los que siguen dando un resultado positivo.
Anemia falciforme
La anemia falciforme o drepanocitosis es una enfermedad hereditaria de los eritrocitos que se trasmite de forma autosómica recesiva.
Es la forma más frecuente de hemoglobinopatía estructural, con un claro patrón étnico, siendo África subsahariana la población con mayor prevalencia mundial. La prevalencia en España varía con la zona geográfica. Para las formas más graves, es de 1/6.000 y para los portadores de 1/500 RN. Se caracteriza por la presencia de hemoglobina S en el hematíe, una forma de hemoglobina inestable que tiende a polimerizarse y ocluir la microcirculación, produciendo manifestaciones multisistémicas, tanto agudas como crónicas. Al nacimiento es asintomática, con aparición de los primeros síntomas hacia los 4-6 meses, cuando comienzan a disminuir los niveles de la hemoglobina fetal (F), en forma de dactilitis o tumoración dolorosa de manos y pies por vasooclusión. La detección precoz de hemoglobinopatías, especialmente para el síndrome drepanocítico, presenta el máximo nivel de evidencia científica y fuerza de recomendación (A1)(15). Sin embargo, la desigualdad en la disponibilidad de recursos hace que la oferta de los PCN no sea aún homogénea entre las diferentes comunidades autónomas.
Se ha demostrado que un diagnóstico precoz junto a la eficacia de la profilaxis antibiótica y la vacunación antineumocócica y frente Haemophilus, puede reducir sustancialmente la morbi-mortalidad durante los 5 primeros años, debido a la disminución de la incidencia de infecciones. También se ha observado que si, además, se incluye una educación de los padres y un seguimiento específico, se contribuye aún más a la reducción de la mortalidad. Además de evitar las muertes prematuras en los RN enfermos, el cribado neonatal permite el adecuado consejo genético y planificación familiar.
Hoy en día, la mayoría de los programas de cribado de hemoglobinopatías utilizan técnicas de Cromatografía Líquida de Alta Resolución (HPLC) o de Isoelectroenfoque (IEF) en eluidos de sangre seca en papel, como métodos de screening. Ambos sistemas tienen una sensibilidad y especificidad muy cercanas al 100% y detectan HbA, F, S, C, E, A2, D-Punjab y O-Arab, entre otras. La HPLC-CE o cromatografía líquida de alta resolución de intercambio catiónico es la técnica más empleada y permite la separación de la HbA de cada una de las variantes según el tiempo de retención característico de cada una de ellas.
La espectrometría de masas en tándem (MS/MS) también puede ser empleada para el cribado de hemoglobinopatías, siendo, de hecho, método de referencia para la detección de variantes de hemoglobina.
Cribado de hipoacusia congénita
El déficit auditivo en la primera infancia dificulta la adquisición del lenguaje, alterando la capacidad de comunicación y aprendizaje del niño y, a largo plazo, su integración social. La incidencia de hipoacusia congénita es alta, afectando a 1-3/1.000 RN vivos.
El 80% de las sorderas infantiles están presentes al nacimiento y el 95% de ellos nacen en familias sin problemas de audición. El cribado limitado a la población de alto riesgo identifica solo al 50% de los RN con hipoacusia, lo que supone que la otra mitad será diagnosticada a una edad tardía, cuando la intervención es menos eficaz.
La realización de la prueba de cribado neonatal auditivo durante la estancia en la maternidad es una medida eficiente, con un claro beneficio si la detección precoz de la sordera moderada a grave se realiza antes de los tres meses de edad. La complejidad y coste de los equipos de cribado hace que sea estratégicamente deseable que el cribado se realice durante los primeros días de vida, mientras el niño está en la maternidad, ya que en España, prácticamente todos los niños nacen en maternidades. Esto, además, permite unificar la metodología y formar adecuadamente al personal que tiene el encargo de realizar la prueba. En los RN grandes prematuros, se realizará un cribado con potenciales auditivos u otoemisiones antes de los tres meses de edad corregida.
Las técnicas objetivas de detección precoz de hipoacusia son las otoemisiones acústicas (OEA) y los potenciales evocados auditivos del tronco cerebral (PEATC). Las OEA se realizan instalando unos minitransductores en el conducto auditivo, que emiten estímulos auditivos y recogen las respuestas sonoras generadas en la cóclea.
La realización de PEATC es la forma de detección precoz más precisa y fiable, aunque más cara. Se colocan tres electrodos en el cuero cabelludo del paciente, que recogen la actividad electrofisiológica generada por estímulos sonoros a diferentes frecuencias.
Como es de esperar en el despistaje de trastornos de baja prevalencia, la mayoría de los casos que no pasan la prueba son falsos positivos. Aunque la implementación del programa requiere un adecuado entrenamiento personal; en España, la Comisión para la Detección Precoz de la Hipoacusia recomienda, desde 1999, el cribado auditivo neonatal(16).
Si el cribado de hipoacusia resulta alterado, se debe establecer el diagnóstico de confirmación de hipoacusia antes del tercer mes de vida. Los niños que precisen re-cribado deben ser evaluados de forma bilateral, aunque en la prueba inicial solo fallara un oído.
Se debe incluir a todo lactante con déficit auditivo confirmado en un programa de intervención multidisciplinar, para establecer el tratamiento indicado antes de los 6 meses de vida, incluyendo intervención logopédica y la eventual adaptación audioprotésica necesaria en cada caso. Por otro lado, se debe controlar periódicamente a todo lactante con factores de riesgo asociados a hipoacusia de aparición tardía, aunque haya pasado las pruebas del cribado neonatal.
Bibliografía
1. Ministerio de Sanidad, Consumo y Bienestar Social. Programas de Cribado neonatal de enfermedades endocrino-metabólicas. Acceso el 15 de enero de 2019. http://www.mscbs.gob.es/profesionales/saludPublica/prevPromocion/cribadoNeonatal.htm
2. Guthrie R and Susi A. A Simple Phenylalanine Method for Detecting Phenylketonuria in Large Populations of Newborn Infants. Pediatrics. 1963; 32: 338-43.
3. Millington DS, Kodo N, Norwood DL, Roe CR. Tandem mass spectrometry: A new method for acylcarnitine profiling with potencial for neonatal screening for inborn errors of metabolism. J Inherit Metab Dis. 1990; 13: 321-4.
4. Wilson JMG, Junger G. Principles and Practice of Screening for Disease. Geneva: World Elath Organization; 1968.
5. Vanderhilst C, Derks T, Reijngoud D, et al. Cost-Effectiveness of Neonatal Screening for Medium Chain acyl-CoA Dehydrogenase Deficiency: The Homogeneous Population of the Netherlands. The Journal of Pediatrics, 2007. 151(2): 115-120.e3.
6. Pandor A, Eastham J, Beverly C, et al. Clinical effectiveness and cost-effectiveness of neonatal screening for inborn errors of metabolism using tandem mass spectrometry: a systematic review. Health Technology Assessment. 2004; 8: 1-121.
7. American College of Medical Genetics Newborn Screening Expert Group. Newborn screening: toward a uniform screening panel and system: executive summary. Pediatrics. 2006; 117(5). www.pediatrics.org/cgi/content/full/117/5/SE1/e296
8. Cornel M, Rigter T, Weinreich S, et al, Newborn screening in Europe Expert Opinion document – Evaluation of population newborn screening practices for rare disorders in Member States of the European Union, E. Comission, Editor. 2012.
9. Almannai M, Marom R, Sutton VR. Newborn screening: A review of history, recent advancements, and future perspectives in the era of next generation sequencing. Curr Opin Pediatr. 2016; 28: 694-9.
10. Lewys MH. Newborn screening controversy: Past, present, and future. JAMA Pediatr. 2014; 168: 199-200.
11. NBS01-A6 Blood Collection on Filter Paper for Newborn Screening Programs; Approved Standard-Sixth Edition, 2013.
12. Rapaport R. Thyroid function in very low birth weight newborn: rescreen or reevaluate. J Pediatrc. 2002; 140: 287-9.
13. Couce ML, Castiñeiras DE, Bóveda MD, Baña A, Cocho JA, Iglesias AJ, et al. Evaluation and long-term follow-up of infants with inborn errors of metabolism identified in an expanded screening programme. Mol Genet Metab. 2011; 104: 470-5.
14. Kim S, Lloyd-Puryear MA, Tonniges TF. Examination of the communication practices between state newborn screening programs and the medical home. Pediatrics. 2003; 111(2). https://pediatrics.aappublications.org/content/111/2/e120.full.
15. Sickle Cell and Thalassaemia. Handbook for Laboratorires. UK: NHS Screening Programmes, 2012.
16. González de Dios J, Mollar Maseres J, Rebagliato Russo M. Evaluación del programa de detección precoz universal de la hipoacusia en el recién nacido. An Pediatr (Barc). 2005; 63: 193-8.
