|
| Temas de FC |
J.C. López Gutiérrez
Servicio de Cirugía Pediátrica. Hospital La Paz. Madrid
Profesor Asociado de Pediatría. Universidad Autónoma de Madrid
| Resumen
La cirugía plástica pediátrica ha avanzado de forma espectacular en las últimas décadas, debido a diversos factores, entre los que destacan: el progreso en secuenciación genómica y análisis del origen genético de la mayoría de las malformaciones congénitas, los adelantos en técnicas diagnósticas cada vez menos invasivas y más eficaces, la sofisticación de las técnicas de anestesia infantil y el desarrollo inexorable de técnicas reconstructivas cada vez más efectivas, complejas y con menos secuelas. |
| Abstract
Pediatric plastic surgery has advanced dramatically in recent decades due to several factors as: progress in genome sequencing and analysis of the genetic origin of most congenital malformations, advances in less invasive and more effective diagnostic techniques, improved safety of pediatric anesthesia procedures and tireless development of progressively effective and less complex tissue reconstruction. |
Palabras clave: Heridas; Nevus; Hemangioma; Quemaduras.
Key words: Wounds; Nevi; Hemangioma; Burns.
Pediatr Integral 2014; XVIII(10): 750-759
Cirugía reparadora pediátrica
Repercusión psicológica en la familia y el niño con malformación congénita
La repercusión psicológica de una malformación grave sobre el entorno familiar es de sobra conocida por los especialistas que tratan a diario a estos pacientes. La presencia de una malformación en cualquier localización y, especialmente, en el territorio facial es el origen de distress emocional llamativo en los padres del lactante, que reaccionan aislándose y adquiriendo comportamientos no habituales.
A su vez, las secuelas desfigurantes postquirúrgicas son el origen de una reducción evidente en la calidad de vida del niño, en el plano educativo, familiar y personal. Trastornos en la aceptación de la imagen corporal, ansiedad, trastornos del sueño y depresión son proporcionales al grado de distorsión que produce la deformidad a largo plazo.
Es evidente que, un tratamiento precoz de la anomalía va a minimizar las futuras secuelas, reduciendo significativamente el impacto psicológico en los padres y sobre todo en el niño.
Es recomendable que los padres de un lactante con malformación severa reciban un seguimiento psicológico periódico y que sean evaluados, precozmente, por un equipo multidisciplinar con experiencia.
Lesiones cutáneas: heridas y quemaduras
El correcto tratamiento de las pérdidas de tejido, sean de origen traumático o térmico, por un equipo experto, reducen significativamente la morbilidad y las secuelas a largo plazo.
Heridas
Las lesiones incisas o contusas en piel son la causa más frecuente de atención quirúrgica urgente en la infancia. La localización más frecuente es la cara y las manos, por lo que dada la importancia estética y funcional deben ser tratadas de forma eficaz y precoz.
La decisión más importante a tomar tras explorar la lesión, es: si la limpieza y sutura de la herida puede hacerse de forma efectiva con anestesia local o si, por el contrario, la reconstrucción adecuada de los tejidos o una exploración minuciosa de estructuras subyacentes deben hacerse bajo anestesia general.
En general, siempre que la herida sea superficial o de pocos centímetros de longitud puede suturarse sin complicaciones bajo anestesia local (mepivacaína 2% preferentemente) usando suturas reabsorbibles (4/0 o 5/0 en tejido subcutáneo para aproximar los bordes y 6/0 para unirlos), con el fin de evitar la dolorosa y molesta retirada de puntos. Si es posible y no hay hemorragia copiosa, es preferible la sutura intradérmica a los puntos sueltos, ya que la cicatriz residual es menor. Si la herida es grande, afecta a zonas importantes, estética o funcionalmente (párpados, nariz, boca, genitales), el sangrado es profuso o se prevé la necesidad de sedación, es preferible derivar al paciente a un centro con atención quirúrgica pediátrica urgente disponible.
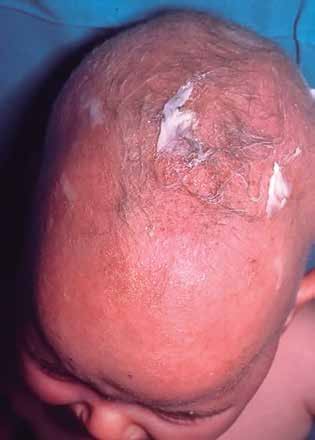
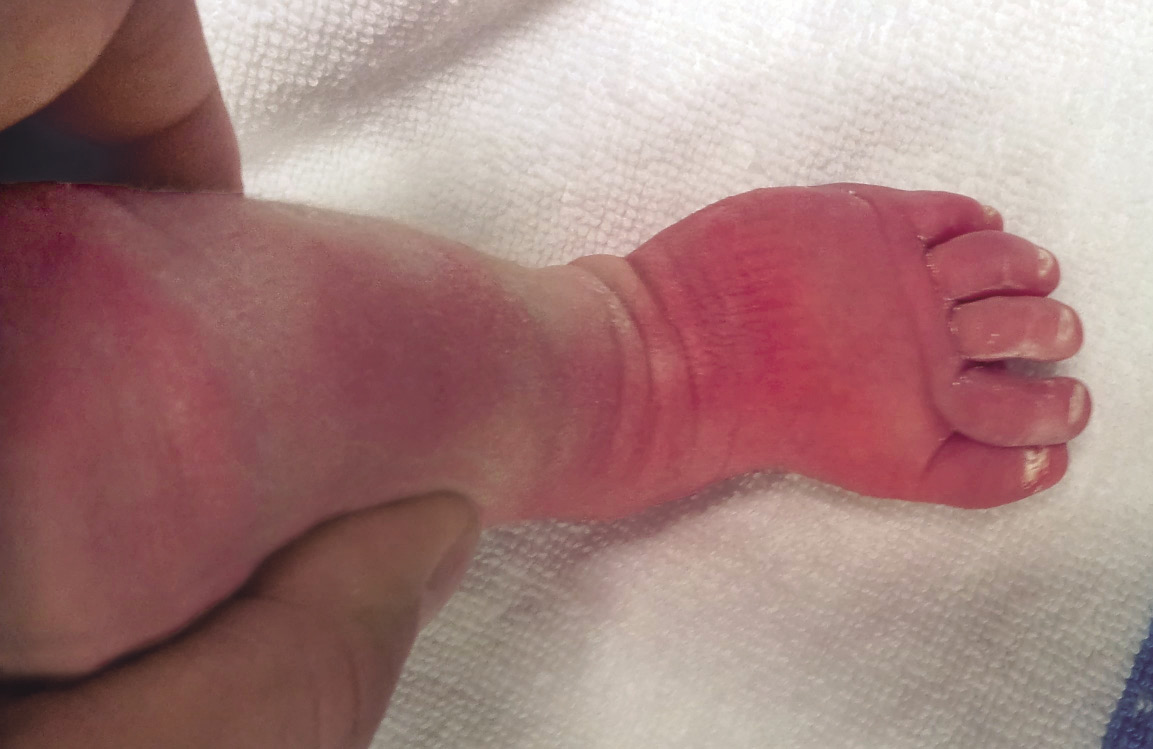
En la actualidad, no se deja abierta una herida bajo ninguna circunstancia incluso en caso de mordedura o infección activa, ya que las cicatrices resultantes son inaceptables (Fig. 1).
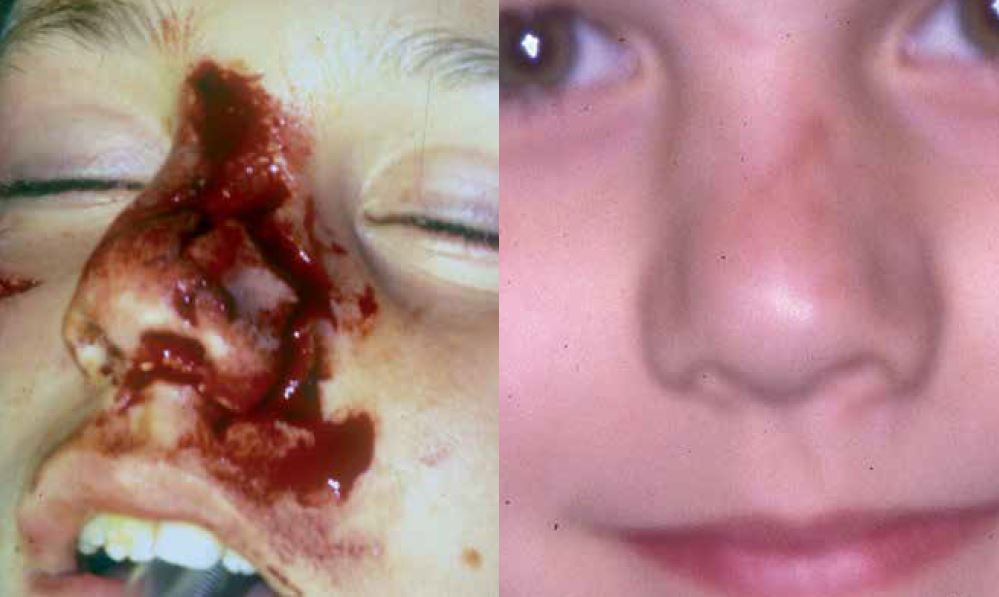
Figura 1. Reconstrucción inmediata de la pirámide nasal, tras mordedura de perro.
Un desbridamiento efectivo y la cobertura antibiótica adecuada, permiten el cierre quirúrgico sin complicaciones. Las heridas profundas en la palma de la mano deben ser revisadas sistemáticamente bajo anestesia general, ya que la lesión de los nervios colaterales y del aparato flexor es la norma. Su reconstrucción debe ser inmediata para evitar la necesidad ulterior de injertos nerviosos o tendinosos.
Si la herida tiene bordes anfractuosos, se ha suturado bajo tensión por pérdida de sustancia, ha habido infección y dehiscencia o no sigue las líneas de Lange de la piel, la cicatrización será hipertrófica en mayor o menor medida, por lo que se recomienda utilizar láminas de silicona tópica autoadhesiva de forma preventiva durante varias semanas y hasta que desaparezca la coloración rosada de la cicatriz.
Las heridas por abrasión que no precisan sutura deben cubrirse con un apósito de silicona en malla y una crema antiséptica, para evitar la adherencia y facilitar las curas indoloras diarias.
Toda herida cuya epitelización completa dure más de 3 semanas producirá cicatrización hipertrófica y debe ser tratada con silicona tópica y/o presoterapia, por lo que debe ser valorada por un cirujano. El tratamiento diferido de la cicatrización hipertrófica es desalentador, por lo que debe hacerse hincapié en la profilaxis.
Quemaduras
Todo niño con quemaduras en extremidades, genitales, cara o en superficie corporal superior al 5%, debe ser ingresado en una unidad de quemados pediátricos.
Las quemaduras siguen siendo la causa de ingresos más prolongados por accidente en la edad pediátrica. Afortunadamente, el pronóstico gracias a los avances en terapia intensiva y quirúrgica, ha pasado de un 50% de supervivencia en quemaduras en el 50% de la superficie corporal hace 40 años a un 98% en la actualidad(1,2).
• Aproximadamente un 30-40% de todos los quemados tiene menos de 15 años, con una edad media de 3 años. Las quemaduras son más frecuentes en varones (65%), en los niños entre 0 y 2 años (15%) y en las clases sociales bajas.
• La cocina es el escenario habitual de las quemaduras en niños pequeños, mientras que los juegos con sustancias inflamables son la causa más frecuente en niños mayores. La incidencia de quemaduras por maltrato sigue aumentando, por lo que deben ser valoradas ante la más mínima sospecha.
• Para prevenirlas, ningún niño debe permanecer sin vigilancia alrededor de un líquido caliente. La modernización de las medidas de protección eléctrica, así como la construcción de viviendas más seguras, están haciendo disminuir drásticamente la incidencia de quemaduras graves.
Tipos de quemaduras
• Las quemaduras térmicas (sobre todo, las escaldaduras) son las más frecuentes (90% de los casos), especialmente en menores de 5 años.
• Las más graves son las producidas por fuego (7% de casos), que predominan entre los 5 y 13 años, y las asociadas a síndrome de inhalación.
• Un 3% de las quemaduras son eléctricas y químicas.
Superficie corporal quemada
Es imprescindible un cálculo muy preciso de la extensión de la piel afectada, por las implicaciones que tiene en la correcta reposición hidroelectrolítica. En el niño menor de 1 año, la cabeza y cuello equivalen al 19% de la superficie corporal total (SCT) y por cada año adicional esta proporción disminuye un 1%; de manera que a los 10 años se iguala con el adulto (9%). Lo mismo ocurre con los miembros inferiores, aumentando un 0,5% anual y llegando al 18% a la edad de 10 años. Los miembros superiores corresponden a un 9% y el tórax y el abdomen a un 36%. La extensión de las quemaduras pequeñas puede ser calculada, utilizando como guía el tamaño de la palma de la mano del propio enfermo, que corresponde aproximadamente al 1% de la SCT. Aunque lo más cómodo y seguro es, siempre, acudir a un nomograma basado en la edad, talla y el peso.
Tratamiento médico
El descenso de la mortalidad está estrechamente ligado a la instauración de equipos multidisciplinares específicos, que incluyen: intensivistas pediátricos, cirujanos, anestesistas, nutricionistas, rehabilitadores y equipos de enfermería entrenados.
Los objetivos del tratamiento de los grandes quemados son, por orden de importancia: preservar la vida, preservar la función, limitar la deformidad física, limitar las secuelas psicológicas y lograr una reintegración social plena.
Indicaciones de ingreso
• Superficie quemada > 5%.
• Quemaduras que afectan a cara, manos o genitales.
Medidas generales
• Evaluación rápida de las lesiones y obtener datos del paciente, como: vacunación, enfermedades asociadas, hora y agente causal del accidente y tratamiento previo recibido.
• Inspección de la mucosa nasal y oral si se sospecha lesión por inhalación. El síndrome de inhalación se produce por la afectación térmica y de productos inhalados del parénquima broncopulmonar, la interrupción del transporte y liberación de oxígeno por los tóxicos inspirados y el daño sistémico por la absorción de sustancias químicas.
• Garantizar una vía aérea permeable. La tráquea del niño quemado es mucho más sensible a la obstrucción por edema. Intubar si existe obstrucción de la vía aérea o el paciente está inconsciente. La intubación supone un importante riesgo de infección en un enfermo severamente inmunodeprimido como es el niño quemado, por lo que la tendencia actual es administrar inicialmente oxigenoterapia con cánulas nasales, e intubar solamente cuando exista hipoxemia o insuficiencia respiratoria progresiva, y no de forma profiláctica.
• Examen del cuerpo en su totalidad, para buscar quemaduras que pudieran haber pasado desapercibidas inicialmente. El pelo próximo a las zonas quemadas debe ser cortado para conocer los límites exactos de la lesión.
• Acceso venoso para reposición hidroelectrolítica y acceso arterial para monitorización. Las vías de elección son: vena periférica en tejido no quemado, vena central en tejido no quemado, vena periférica en tejido quemado y vena central en tejido quemado, por este orden de preferencia. En caso de dificultad para el acceso, la vía intraósea está indicada para el inicio de la reanimación. Existe un alto porcentaje de complicaciones relacionadas con catéteres centrales en pacientes quemados, por la alta susceptibilidad a infecciones y a episodios embólicos secundarios al estado de hipercoagulabilidad.
• Analgesia. Es necesaria en todos los casos. Debe evaluarse la nocicepción con escalas de valoración clínica, especialmente, en menores de 5 años. El analgésico utilizado dependerá de la edad del niño y del tipo de quemadura o procedimiento. Habitualmente se emplea:
- Metamizol: 0,1 ml (40 mg)/kg/6-8 horas por vía iv o vo.
- Meperidina: 0,5 -1 mg/kg/6-8 horas, vía iv o im.
- Cloruro mórfico: 0,1-0,2 mg/kg/3-8 horas vía iv, sc u oral. De elección por varios motivos: eficaz, utilizable por todas las vías y farmacocinética conocida (inicio rápido y larga duración).
- En niños mayores de 5 años, se puede utilizar analgesia controlada por el paciente con perfusión continua de mórficos.
- Los cambios de apósito deben hacerse, previa administración profiláctica de analgésicos, media hora antes del procedimiento.
• Hay que tener en cuenta que la causa más frecuente de agitación e inquietud en el quemado es la hipoxia cerebral, bien por hipoxemia o por hipovolemia, por lo que debe asegurarse una buena oxigenación y perfusión periférica.
• Sonda nasogástrica o nasoyeyunal para inicio precoz de la nutrición enteral. En quemaduras faciales o extensas [Superficie Corporal Quemada (SCQ) > 50 %], se puede realizar una gastrostomía endoscópica percutánea a las 48 horas, para mayor comodidad del niño.
• Protección gástrica.
• Control de la diuresis mediante sondaje vesical. La diuresis deberá mantenerse entre 0,5-2 ml/kg/h.
• Tratamiento del síndrome catabólico, responsable de la pérdida masiva de peso: se valora según la gravedad de las quemaduras, el uso de hormona de crecimiento, oxandrolona, insulina y propranolol.
Reposición hidroelectrolítica
Los objetivos de la reposición de líquidos y electrolitos son: reposición de pérdidas secundarias a la quemadura, mantener los requerimientos basales, minimizar la formación de edema, normalizar el equilibrio ácido-base, restaurar el nivel de electrolitos y proteínas a valores normales, y conseguir una perfusión adecuada.
• Rehidratación en las primeras 24 horas: la estimación del volumen a infundir, viene determinada por la extensión y profundidad de la quemadura, por el peso del paciente y por la hora exacta del accidente.
Existen numerosas fórmulas para el cálculo de la administración de líquidos en el niño, con quemaduras durante las primeras 24 horas. Las más fiables son:
• Fórmula de Galveston: 5.000 ml/m2 SCQ + 2.000 ml/m2 SCT (basales).
• Fórmula de Parkland: 3-4 ml/kg x % SCQ + necesidades basales.
Ritmo de infusión:
Administrar la mitad de lo calculado en las primeras 8 horas postquemadura y la otra mitad en las siguientes 16 horas.
Aunque la administración de fluidos se realice según las pautas mencionadas, el volumen y ritmo de infusión se irán variando según la respuesta del paciente, la diuresis y el ionograma.
Fluidos:
• Cristaloides en forma de Ringer en las primeras 24 horas. Se prefiere el Ringer, porque su pH de 6,5 es más fisiológico que el del suero salino, pH de 5. Puede enriquecerse con sodio hasta los 180 mEq/L para disminuir el volumen de los aportes, vigilando los niveles séricos de electrolitos.
• También se pueden utilizar soluciones salinas hipertónicas (400-600 mOsm/L). Son eficaces para tratar el shock, porque favorecen el paso de agua desde el espacio intersticial al vascular, limitando la formación de edema, aumentan la contractilidad miocárdica y disminuyen las resistencias vasculares. En contraposición pueden producir hiperosmolaridad, hipernatremia, deshidratación celular, hemorragia cerebral o edema, e inhibición de la lipolisis.
• Coloides como albúmina o plasma. Las proteínas plasmáticas son imprescindibles para el mantenimiento de la presión oncótica en el espacio vascular. En las quemaduras, la pérdida de proteínas es muy alta durante las primeras 6 horas, por lo que es aconsejable no utilizarlas antes de este tiempo, ya que se pierden rápidamente por la elevada permeabilidad vascular en las zonas lesionadas. En la mayoría de los casos se utiliza albúmina (12,5 g por litro de Ringer) para iniciar el aporte proteico.
- Segundo día: 3.750 ml/m2 SCQ + 1.500 ml/m2 SCT.
Infección
Las quemaduras se colonizan por enterobacterias precozmente. Si no se inicia la alimentación enteral de forma inmediata, la infección profundiza e invade un tejido sano y viable, agravando el pronóstico. Las medidas más eficaces en la prevención de la infección son:
1. Limitar al máximo las puertas de entrada (cuidado de catéteres, retirada precoz del tubo endotraqueal, etc.).
2. Cirugía precoz y cierre definitivo de la herida lo antes posible.
3. Curas diarias con lavado de las lesiones y aplicación de apósitos específicos o en su defecto crema de sulfadiazina argéntica.
4. Soporte nutricional agresivo y precoz.
5. Administración de antibióticos solo cuando estén indicados: sepsis, condritis, infección nosocomial y estados de inmunosupresión. Los antibióticos sistémicos se utilizan solo para tratar las infecciones establecidas y nunca profilácticamente, excepto en dos situaciones:
• Pacientes de alto riesgo de infección por Streptococcus beta hemolítico (portador o exposición reciente al mismo): profilaxis antibiótica con dosis bajas de penicilina.
• En las primeras 24 horas previas y subsecuentes a la escisión de las lesiones, para proteger al paciente de los efectos de la bacteriemia transitoria.
La antibioterapia se realiza según los cultivos y antibiogramas. En profilaxis peroperatoria, se emplea una asociación de cefalosporina (cefazolina: 50-100 mg/kg/día en 3-4 dosis) y aminoglucósido (tobramicina o gentamicina: 5 mg/kg/día en 1 a 3 dosis).
Metabolismo y nutrición
• El objetivo final es que el niño tenga el mismo peso al alta, que el día del ingreso.
• Inicio de la nutrición: la lesión térmica produce un marcado grado de hipermetabolismo, por lo que la nutrición se debe iniciar precozmente, a las 2 horas de la quemadura.
• Vía de administración: siempre que sea posible debe utilizarse la vía enteral (oral, nasogástrica o yeyunal). Si el niño no está consciente, se utilizará una sonda transpilórica o gastrostomía endoscópica. La nutrición parenteral aumenta el riesgo de mortalidad en los grandes quemados y debe limitarse al mínimo imprescindible.
• Aporte calórico: debe ajustarse diariamente controlando el balance nitrogenado, el peso y la calorimetría indirecta. En quemados de >20% de la SCT, se puede emplear la fórmula de Galveston:
1.100 al/m2/día de SCT + 1.300 kcal/m2/día (en niños entre 2-15 años).
2.100 kcal/m2/día de SCT + 1.000 kcal/m2/día de SCQ (en menores de 2 años).
• Tipo de nutriente y ritmo de alimentación: es recomendable que un especialista en nutrición pediátrica supervise el aporte energético, desde el día del accidente hasta el alta hospitalaria.
Como norma general se usan:
• En menores de 2 años: hidrolizado de proteínas. Inicio a 2 ml/kg/h, subiendo 1 ml/kg/h cada día, hasta llegar a 5 ml/kg/h.
• En mayores de 5 años: fórmulas enterales completas. Impact® (dieta enriquecida con arginina, RNA y ácidos grasos omega 3) en superficies menores del 30% y Alitraq® (dieta elemental suplementada con glutamina) en mayores del 30%. Inicio a 10-20 ml/h, subiendo 10 a 30 ml/día, hasta alcanzar las necesidades calóricas calculadas. El 4º día se inicia la alimentación normal del niño y la enteral pasa a ser exclusivamente nocturna.
• La utilización de oxandrolona a 0,1 mg/kg/12 h durante al menos una semana, pamidronato durante un mes y hormona de crecimiento recombinante a dosis bajas durante un año, mejoran los parámetros endocrino-metabólicos en niños con quemaduras graves.
Tratamiento quirúrgico
El primer paso del tratamiento es neutralizar el origen de la quemadura.
Quemadura térmica: el primer objetivo consiste en detener el proceso térmico. Por lo tanto, es indispensable retirar pronto todas las ropas (la ropa adherida debe ser eliminada después del ingreso, cuando se realice la limpieza y desbridamiento de la lesión) e inmediatamente aplicar agua fría durante al menos 5 minutos. En ningún caso debe aplicarse hielo, ni cualquier otro tipo de sustancias. Esta simple medida puede hacer que las quemaduras sean superficiales y no necesiten injerto cutáneo.
Quemaduras químicas: es urgente eliminar el agente químico. Las quemaduras por ácidos deben ser irrigadas durante unos 30 minutos bajo chorro de agua.
Recubrimiento con apósitos: los más usados son Acticoat®, Aquacel Ag®, Suprathel, Biobrane® y E-Z Derm®. Tiene como ventajas:
• Reducir el número de curas, siendo más cómodo para los niños.
• Disminuye el dolor, el sangrado (al ser menor el número de curas) y la pérdida de líquidos.
• Delimita las zonas de epitelización espontánea (dermis superficial), de las que precisarán autoinjerto 5 o 7 días después (dermis profunda).
• Por su adaptabilidad pueden ser usados en cualquier área del cuerpo incluida la cara.
• La cobertura con membrana amniótica puede ser eficaz y con mejores resultados a largo plazo en quemaduras faciales dérmicas no profundas.
Escarotomías: son incisiones profundas hasta la fascia, que se practican cuando hay quemaduras circulares con disminución de la perfusión distal, especialmente, en extremidades, tórax y cuello. La incisión debe abarcar todo el espesor de la piel quemada, para que el espacio que se encuentra debajo pueda expandirse, disminuyendo la presión tisular. La elevación inmediata de la extremidad quemada disminuye el edema. La perfusión se controla con pulsioxímetro.
Desbridamiento: actualmente se realiza una escarectomía o desbridamiento precoz (en las primeras 48 horas), evitándose así una gran pérdida sanguínea y una reacción inflamatoria masiva. Se ha demostrado que una escarectomía en las primeras 24 horas, supone una pérdida sanguínea de 0,4 ml/cm2, que es similar a la pérdida hemática de las curas tras 15 días con tratamiento tópico.
Injertos:las quemaduras superficiales curan espontáneamente en 2 semanas sin dejar apenas cicatrices. Las quemaduras profundas precisan escarectomía tangencial y autoinjerto, tan pronto como sea determinada su profundidad y el estado hemodinámico del niño lo permita. El injerto obtenido de piel parcial puede mallarse (1:1,5 o 1:3) y de esta forma cubrir una mayor superficie, pero es desaconsejable y debe evitarse en quemaduras de poca extensión, ya que el resultado cosmético es significativamente peor. Cuando por la amplia extensión de las lesiones no se dispone de suficientes autoinjertos, se recurre a sustitutos de piel. El más utilizado es la piel de cadáver criopreservada, que puede permanecer varias semanas adherida al lecho de la quemadura evitando la infección, hasta que se lleve a cabo el autoinjerto.
Recientemente se ha extendido el uso de regeneradores dérmicos, que tienen la misma función que la piel de cadáver, pero con menor riesgo de transmisión vírica, no hay que retirarlos y consiguen mejor resultado estético.
En grandes quemados (>70% SCQ) se utilizan queratinocitos cultivados. En este caso, se toma una biopsia de piel inmediatamente y se manda al laboratorio para cultivo. Esta biopsia se obtiene de una zona sana y de un tamaño aproximado de 2 x 2 cm. A las 3 semanas de su cultivo, se dispone de la suficiente piel cultivada como para recubrir toda la superficie quemada del paciente. El reto, por tanto, está en conseguir la supervivencia del paciente durante este tiempo, manteniendo el lecho de la herida limpio y bien vascularizado, para que el cultivo prenda de forma óptima.
Tratamiento de las secuelas
La rehabilitación debe iniciarse a los pocos días del tratamiento quirúrgico con fisioterapia intensiva y ferulización activa y pasiva.
La cicatrización hipertrófica se combate con presoterapia y silicona tópica.
El apoyo psicológico al niño y a los padres debe iniciarse lo antes posible y prolongarse hasta la recuperación completa.
La deprivación del sueño y el prurito son secuelas frecuentes que hay que vigilar y tratar.
Toda alteración funcional o estética debe ser tratada quirúrgicamente de forma precoz, mediante plastias, injertos y colgajos locales o a distancia.
Nevus congénitos
El tratamiento quirúrgico de los nevus grandes debe ser consensuado por la familia, el dermatólogo y el cirujano. Una vez que se ha decidido indicarlo, no hay ningún beneficio en esperar a edades tardías.
Según datos del Nevus Network, asociación de pacientes con más de 1.000 casos censados de nevus congénitos gigantes, la tasa de mortalidad por malignización a melanoma sigue reduciéndose, llegando a ser casi inexistente para nevus solitarios y uniformes, aunque sean de gran tamaño. Siendo, pues, el riesgo de degeneración pequeño, la indicación prevalente para la extirpación es cosmética, lo cual hace que la decisión pueda posponerse varios años o incluso indefinidamente. Esto no es óbice para que, el equipo dermatológico-pediátrico-quirúrgico sea consciente de que algunos pacientes son candidatos a un tratamiento quirúrgico temprano, basado en las características clínicas, dermatoscópicas, sociales o familiares(3,4).
El momento idóneo para la extirpación quirúrgica de nevus melanocíticos congénitos sigue siendo objeto de controversia entre pediatras, dermatólogos, cirujanos y familiares.
La indicación oncológica de extirpación preventiva de estas lesiones es siempre discutible, dada la bajísima tasa de malignización de las mismas. Sin embargo, la indicación cosmética sigue manteniéndose inalterable y pocos son los niños que, una vez reconocida su imagen corporal hacia los 5 o 6 años, no muestran rechazo frontal hacia una tumoración pigmentaria grande, cualquiera que sea su localización.
La extirpación quirúrgica por motivos cosméticos cambia las expectativas de la familia y el resultado debe ser no solo eliminar la “mancha” sino conseguir una cicatriz poco perceptible. En nombre del riesgo oncológico, valen resultados estéticos que son inaceptables cuando la indicación es cosmética (por ejemplo, las cicatrices resultantes del uso de injertos laminares). Establecer la indicación quirúrgica en el recién nacido o lactante es excepcional. Sin embargo, un protocolo sensato de evaluación de lesiones pigmentarias por una unidad médico-quirúrgica debe incluirlas, pues como lo demuestra nuestra experiencia, se presentan ocasionalmente.
La adecuación de la técnica al tamaño y localización del nevus es el punto determinante para la obtención de un buen resultado estético y funcional. La experiencia juega en estos casos un factor determinante para conseguir los objetivos previstos por la familia, el dermatólogo y el propio cirujano de forma conjunta. La explicación detallada del procedimiento, incluyendo imágenes de casos similares, así como la entrevista de los padres con familiares que han pasado por un proceso similar son de ayuda inestimable cuando hablamos de intervenciones técnicamente difíciles en niños de corta edad.
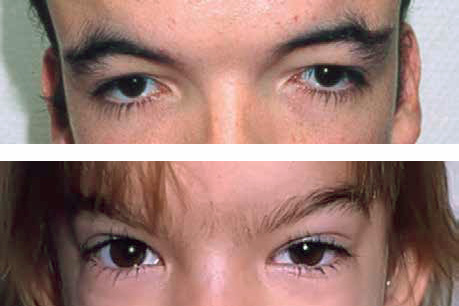
De cualquier manera, es de consenso entre las unidades especializadas a nivel internacional, que la ausencia de riesgo y la calidad superior de los resultados son la norma a favor de la extirpación precoz frente a la extirpación tardía (Fig. 2). Esta última añade a los factores intercurrentes repercusión psicológica y absentismo escolar, que hacen habitual, entre los pacientes de este grupo, la queja o desazón por no haber llevado a cabo la intervención a una edad más temprana.
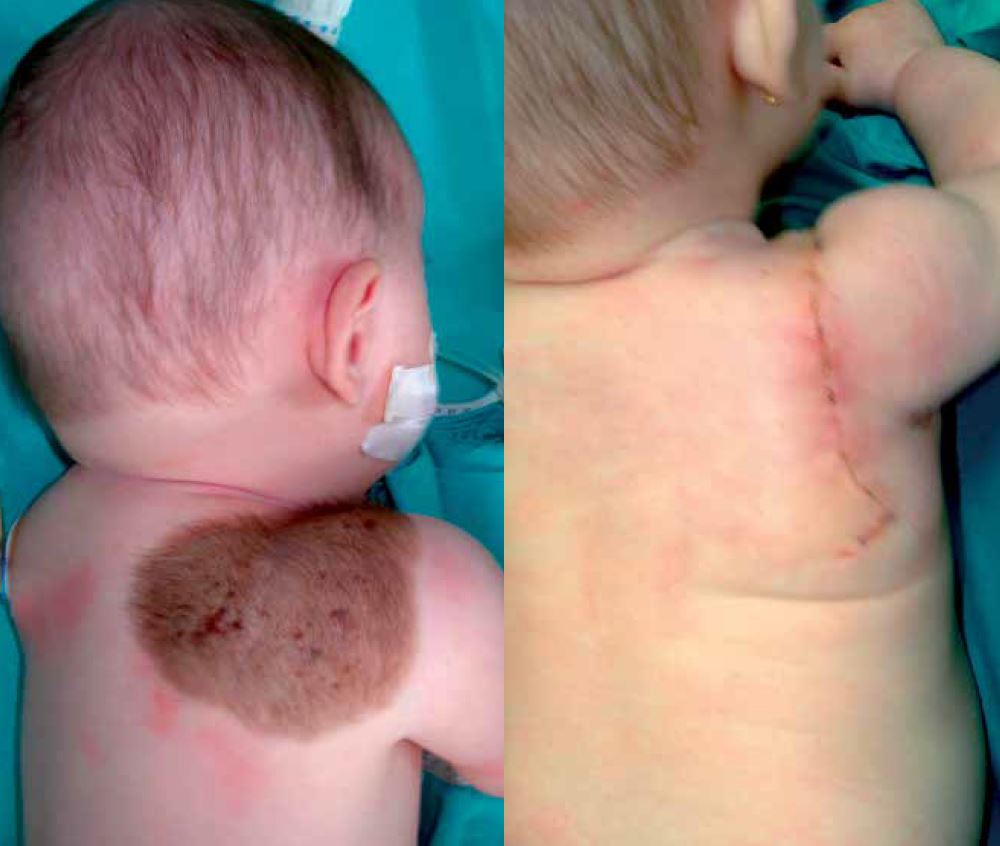
Figura 2. Nevus Congénito. Extirpación y cierre primario en un solo tiempo. Esta técnica y resultado solo pueden conseguirse si la intervención se realiza durante el primer año de vida.
En resumen, creemos que la selección adecuada de los candidatos a extirpación precoz debe ser realizada por un equipo multidisciplinar con experiencia en dermatología y cirugía reconstructiva pediátrica, la cual es imprescindible para mejorar el grado de satisfacción de la familia y a largo plazo del niño(5,6).
Hemangiomas
Todo niño que presenta un hemangioma en cara y con crecimiento llamativamente rápido en el primer mes de vida debe ser tratado con beta-bloqueantes para evitar tratamiento quirúrgico posterior.
Si consideramos que hasta un 25% de los recién nacidos pretérmino con un peso inferior a 1.000 g presentan hemangiomas y que este grupo de pacientes sigue incrementándose a medida que avanzan las técnicas de fecundación in vitro, es evidente que nos encontramos ante un fenómeno epidemiológicamente relevante.
Los hemangiomas han rebasado ya hace tiempo el trasnochado criterio de no ser tratados, porque involucionan espontáneamente, y demandan de los especialistas involucrados mayor comunicación para establecer pautas terapéuticas más efectivas. El fenómeno de involución no implica desaparición completa de todo rastro del tumor. La ulceración deja, sistemáticamente, una cicatriz indeleble y el residuo fibrograso que perdura, una vez que desaparece la estructura vascular de la lesión, precisará extirpación quirúrgica o lipoaspiración.
Los betabloqueantes y concretamente el propranolol a 3 mg/kg/día, se han convertido en el fármaco de elección en el tratamiento de los hemangiomas infantiles, no solo cutáneos sino viscerales y en vía aérea(7,8).
En España, la confianza en esta nueva indicación terapéutica ha sido evidente desde el descubrimiento de su eficacia y la lista de unidades que lo utilizan de forma habitual, así como el número de especialistas de todas las áreas de la medicina que lo prescriben, va en aumento.
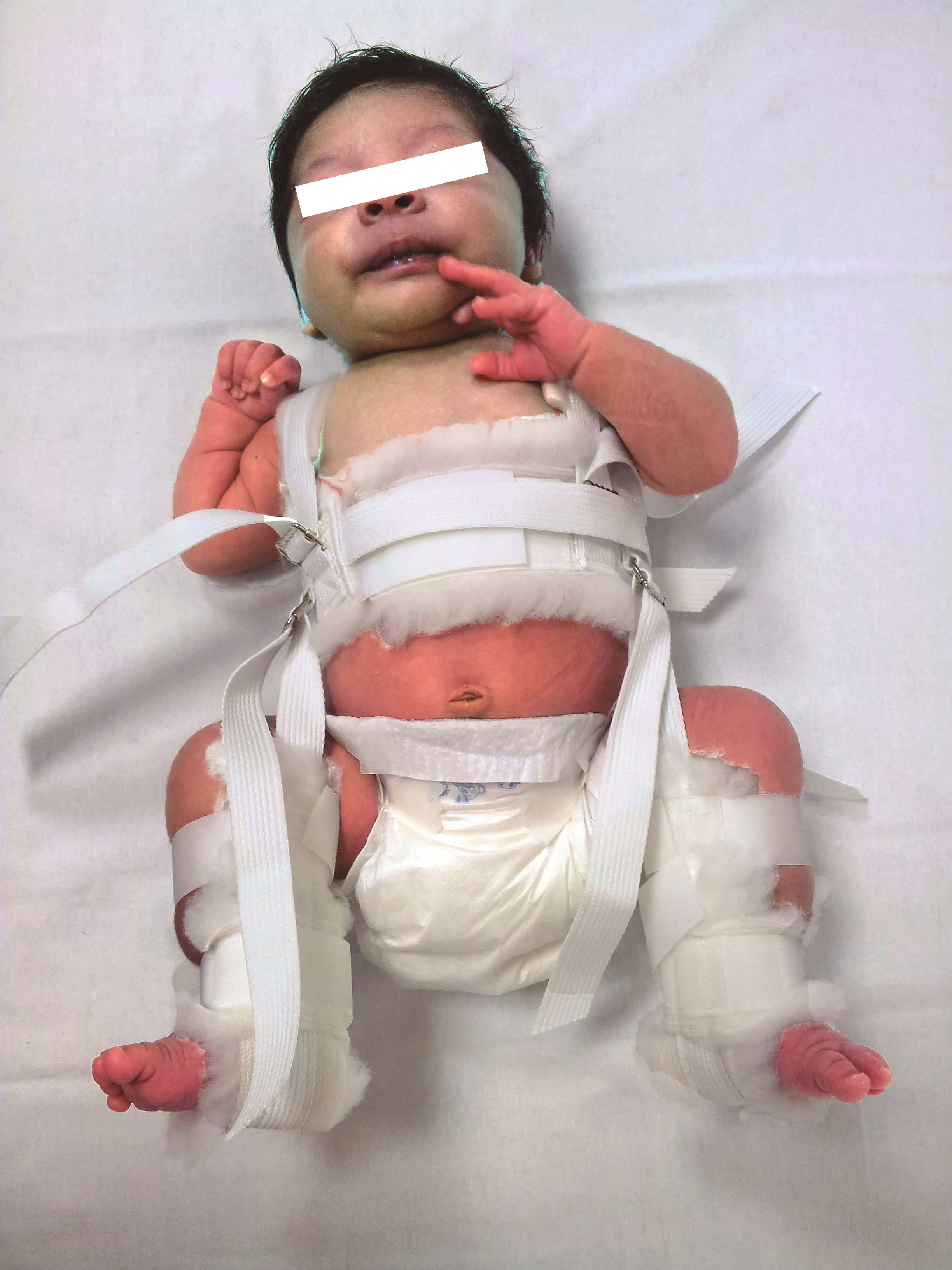
El resultado está siendo ya, una reducción drástica en el número de intervenciones quirúrgicas –que, en algunos casos, incluían el trasplante hepático– y una disminución en la tasa de complicaciones visuales y respiratorias. Además, las frecuentes ulceraciones han pasado a ser un hecho excepcional y de solución rápida (Fig. 3).

Figura 2. Hemangioma ulcerado. Respuesta inmediata al tratamiento con propranolol a 3 mg/kg/día.
A pesar de todos estos avances, todavía debemos seguir trabajando por conocer exactamente los mecanismos de acción y, en función de estos, el tratamiento farmacológico de otras anomalías vasculares.
El propranolol, como uno de los mayores éxitos del creciente fenómeno de “drug repositioning”, está llamado a liderar el descubrimiento de nuevos fármacos con poder antiangiogénico(9,10).
Malformaciones linfáticas
Es imprescindible identificar correctamente el tipo de malformación linfática, antes de tomar ninguna decisión terapéutica.
A pesar de los importantes avances logrados en el conocimiento de las anomalías vasculares, la terminología incorrecta continúa impregnando la comunidad médica, lo que aumenta la probabilidad de que un paciente reciba un tratamiento incorrecto. Este hecho, frecuente entre los tumores vasculares y las malformaciones venosas, arteriovenosas y capilares, felizmente es excepcional en las malformaciones linfáticas (ML). Solo el término hemolinfangioma, aún por erradicar, establece dificultad diagnóstica entre malformaciones mixtas linfático-venosas y malformaciones linfáticas con sangrado intralesional.
En un principio las ML se incluyeron entre las malformaciones de bajo flujo, pero dadas las múltiples dificultades en el manejo de muchos pacientes con malformaciones linfáticas complejas y compromiso de distintos órganos, así como la presencia de componente linfático en ciertos tumores, se reclasificaron en tumores (entre los que se incluirían el hemangioendotelioma kaposiforme y el linfangiosarcoma) y en malformaciones linfáticas [que comprenden las malformaciones linfáticas comunes (lesiones localizadas macroquísticas y microquísticas), la malformación linfática generalizada (MLG), el síndrome de Gorham– Stout, la linfangiomatosis kaposiforme (LK), las anomalías de los canales de conducción y el linfedema].
En la actualidad, el progresivo conocimiento de la fisiopatología de las malformaciones linfáticas, su más conveniente clasificación y los adelantos técnicos en su tratamiento, tanto quirúrgicos como endovasculares, han permitido mejorar considerablemente los resultados a largo plazo de los pacientes con las anomalías más complejas y minimizar las secuelas(11-14).
Pueden detectarse en época prenatal, perinatal, manifestarse en la infancia, la adolescencia y durante la vida adulta. Los síntomas pueden ser moderados o severos. Estas afecciones rara vez remiten sin algún tipo de intervención terapéutica y pueden poner en riesgo la vida del paciente.
Desde el punto de vista diagnóstico, la resonancia magnética confirma el origen y la extensión de la lesión.
Desde el punto de vista terapéutico, la decisión debe ser consensuada por un equipo quirúrgico-radiológico que decida si es mejor la esclerosis o el tratamiento excisional, en función del tamaño, localización y edad del paciente.
Orejas procidentes
La procidencia auricular es un defecto leve que produce, sin embargo, una significativa repercusión psicológica y, por tanto, un trastorno en el desarrollo escolar de los niños con afectación severa. Por este motivo, la corrección de la deformidad es motivo frecuente de consulta en las unidades de cirugía infantil. Esta puede realizarse a cualquier edad, pero se aconseja no demorar hasta la pubertad un procedimiento que va a ser realizado de cualquier forma y que llevado a cabo de forma precoz, minimiza las secuelas psicológicas, no habiendo ventaja alguna en realizarlo en edades tardías.
Sindactilia
Esta anomalía de separación de los dedos es la malformación más frecuente de la extremidad superior y puede ser simple, cuando solo la piel une a los dedos, o compleja cuando hay fusión ósea. La corrección suele realizarse antes del año de edad en las formas graves y un poco más tarde (12-15 meses) en las formas leves.
Anomalías de la mama
La ginecomastia severa y la hipoplasia mamaria en el síndrome de Poland, son los trastornos que más frecuentemente necesitan corrección quirúrgica en la edad pediátrica.
La mamoplastia de reducción está indicada en todo niño con repercusión psicológica por la deformidad.
La simetrización mamaria en el síndrome de Poland tiene indicaciones y timing diferentes en varones y mujeres. En ambos hay que tener en cuenta el defecto de la pared torácica si lo hubiere.
En los niños, la corrección se lleva a cabo en edades más tempranas, utilizándose: injerto autólogo de grasa, colgajo muscular de dorsal ancho, epiplón o material protésico.
En las niñas, además, será necesaria una prótesis definitiva previo implante de un expansor temporal durante todo el periodo de desarrollo mamario(15,16).
Respecto a la mamoplastia de aumento en niñas sin malformación subyacente, la controversia es ilimitada y varios aspectos importantes como: las indicaciones, la edad y la financiación, siguen siendo discutidos.
El primer problema es que fijar la madurez psicológica en los 18 años, va en contra de la propia legislación. Los menores maduros pueden tomar decisiones respecto a someterse o no a intervenciones quirúrgicas y legalmente, cuando la paciente con hipoplasia mamaria tiene más de 16 años, puede decidir por sí misma, aunque sus padres no lo aprueben.
Es extraordinariamente complejo garantizar que una menor con hipoplasia leve o moderada, vea mejorada su vida social y su estado psicológico por el implante. Cuando los progenitores están de acuerdo, no hay problema, pero en numerosas ocasiones la hija quiere operarse y los padres no. En estos casos, la ley ampara al menor y le otorga el derecho a decidir. Sin embargo, las operaciones suelen estar financiadas por los padres, ya que la sanidad pública no las incluye en su catálogo de prestaciones y, por tanto, ellos tienen la última palabra. De este conflicto, solo puede salirse mediante una discusión serena, no limitada a una sola consulta, que incluya una valoración multidisciplinar por un paidopsiquiatra experto, el pediatra y el cirujano.
Malformaciones de la nariz
Las secuelas del contorno nasal en pacientes fisurados son las alteraciones que más frecuentemente requieren corrección quirúrgica en la edad pediátrica. La corrección cosmética del contorno del dorso nasal implica una remodelación ósea, que solo conviene realizar una vez que se haya completado la maduración de la pirámide nasal, lo cual no ocurre antes de los 16 años.
Bibliografía
Los asteriscos reflejan el interés del artículo a juicio del autor.
1. Meuli M, Lochbühler H. Current concepts in pediatric burn care: General management of severe burns. Eur J Pediatr Surg. 1992; 2: 195-200.
2. Eadie PA, Williams R, Dickson WA. Thirty five years of paediatric scalds: are lesson being learned? Br J Plast Surg. 1995; 48: 103-105.
3. Bett BJ. Large or multiple congenital melanocytic nevi: occurrence of neurocutaneous melanocytosis in 1008 persons. J Am Acad Dermatol. 2006 May; 54(5): 767-77.
4. Bett BJ. Large or multiple congenital melanocytic nevi: occurrence of cutaneous melanoma in 1008 persons. J Am Acad Dermatol. 2005 May; 52(5): 793-7.
5.** Bauer BS, Corcoran J. Treatment of large and giant nevi. Clin Plast Surg. 2005 Jan; 32(1): 11-8.
6. Arneja JS, Gosain AK. Giant congenital melanocytic nevi. Plast Reconstr Surg. 2007 Aug; 120(2): 26e-40e.
7.*** Leaute-Labreze C, Dumas de la Roque E and Hubiche T et al., Propranolol for severe hemangiomas of infancy, N Engl J Med 358 (2008), pp. 2649–2651.
8. Sans V, Dumas de la Roque E and Berge J et al., Propranolol for severe infantile hemangiomas: follow-up report, Pediatrics 124 (2009), pp. 423–431.
9. Lawley LP, Siegfried E and Todd JL, Propranolol treatment for hemangioma of infancy: risks and recommendations, Pediatr Dermatol 26 (2009), pp. 610–614.
10. Frieden IL and Drolet BA, Propranolol for infantile hemangiomas: promise, peril, pathogenesis, Pediatr Dermatol 26 (2009), pp. 642–644.
11. Smith MC, Zimmerman B and Burke DK et al., Efficacy and safety of OK-432 immunotherapy of lymphatic malformations, Laryngoscope 119 (2009), pp. 107–115.
12. Burrows PE, Mitri RK and Alomari A et al., Percutaneous sclerotherapy of lymphatic malformations with doxycycline, Lymphat Res Biol 6 (2008), pp. 209–216.
13. Nehra D, Jacobson L and Barnes P et al., Doxycycline sclerotherapy as primary treatment of head and neck lymphatic malformations in children, J Pediatr Surg 43 (2008), pp. 451–460.
14. Alomari AI, Karian VE and Lord DJ et al., Percutaneous sclerotherapy for lymphatic malformations: a retrospective analysis of patient-evaluated improvement, J Vasc Interv Radiol 17 (2006), pp. 1639–1648.
15. Cerrato F, Webb ML, Rosen H, Nuzzi L, McCarty ER, DiVasta AD, Greene AK, Labow BI. The impact of macromastia on adolescents: a cross-sectional study. Pediatrics. 2012 Aug; 130(2): 339-46.
16. Kulkarni D, Dixon JM. Congenital abnormalities of the breast. Womens Health (Lond Engl). 2012 Jan; 8(1): 75-86.
| Caso clínico |
|
Paciente de 5 meses de edad, con tumoración de crecimiento progresivo en el surco nasogeniano y canto orbitario interno izquierdos. Comenzó hace 2 semanas, con el tamaño de un guisante y actualmente tiene 3 cm de diámetro, comienza a comprimir el globo ocular y a obstruir el campo visual, por lo que la paciente acude a consulta. A la exploración, la masa es elástica con leve enrojecimiento cutáneo, sin latido significativamente palpable, cierto aumento de la temperatura e indolora a la exploración. El resto de la exploración y anamnesis no aportan datos de interés, por lo que se solicita una ecografía cuyo informe concluye: masa redondeada en la localización descrita, de bordes bien delimitados con intensa vascularización de forma uniforme y sin contenido quístico. |
Puede realizar el cuestionario de acreditación pinchando AQUÍ.
Le recordamos que debe ser socio y estar registrado.
El cuestionario debe realizarlo en el orden en que los temas aparecen en la revista (del primero al último).
Al finalizar las preguntas de cada tema, estas se guardan automáticamente.