|
| Temas de FC |
A. Farran*, M. Illan**, L. Padró***
*Biólogo, experto en tablas de composición de alimentos. Profesor del Campus de la Alimentación de la Universidad de Barcelona. **Dietista-Nutricionista y Antropóloga. Profesora del Campus de la Alimentación de la Universidad de Barcelona.
***Dietista-Nutricionista, Máster en Educación para la Salud. Profesora jubilada del Campus de la Alimentación de la Universidad de Barcelona
| Resumen
La alimentación saludable es aquella que permite mantener un óptimo estado de salud, cubriendo las necesidades nutricionales para el desarrollo y conservación del organismo y que responde a los conceptos de suficiencia, equilibrio, variedad y adaptación a cada situación y circunstancia. |
| Abstract
Healthy eating is that which is based on the individual needs of each person and which meets the criteria of being: sufficient, balanced, varied and adapted to each and every situation and circumstance. |
Palabras clave: Alimentación vegetariana; Infancia; Adolescencia; Biodisponibilidad; Guías Alimentarias
Key words: Vegetarian diet; Children; Adolescence; Bioavailability; Dietary Guidelines
Pediatr Integral 2015; XIX (5): 313-323
Dieta vegetariana y otras dietas alternativas
Introducción
La alimentación saludable es aquella que permite un óptimo desarrollo durante la infancia y el mantenimiento de la salud.
Una alimentación adecuada debe permitir un óptimo crecimiento y desarrollo durante la infancia, el mantenimiento de la salud, la actividad en la edad adulta y la supervivencia y el confort en la vejez.
La elección de los alimentos se convierte, en la práctica cotidiana, en el acto de comer. Este acto voluntario de ingerir alimentos y combinarlos en los diferentes platos y tomas que realizamos a lo largo del día obedece a las normas que las distintas civilizaciones y culturas han ido generando a lo largo de la historia y configuran el patrón alimentario de los distintos grupos de individuos que conforman la sociedad actual. Teniendo en cuenta todas estas implicaciones, podemos decir que comer es, para los humanos, algo más que alimentarse.
En este contexto se describe, hoy en día, la alimentación saludable y que responde al amplio criterio de ser suficiente, equilibrada, variada y adaptada.
• Suficiente en energía y nutrientes (según edad, sexo, actividad, situación fisiológica…).
• Equilibrada atendiendo a las proporciones recomendadas (55% del valor energético total del día (VET) en forma de hidratos de carbono, 30% VET de lípidos y 15% VET de proteínas).
• Variada para facilitar el aporte de los macro y micronutrientes.
• Adaptada a las condiciones geográficas, culturales, religiosas e individuales(2).
Las Guías Alimentarias
Las Guías Alimentarias para la población española son un conjunto de documentos que informan sobre las normas que se deben seguir para realizar una alimentación saludable.
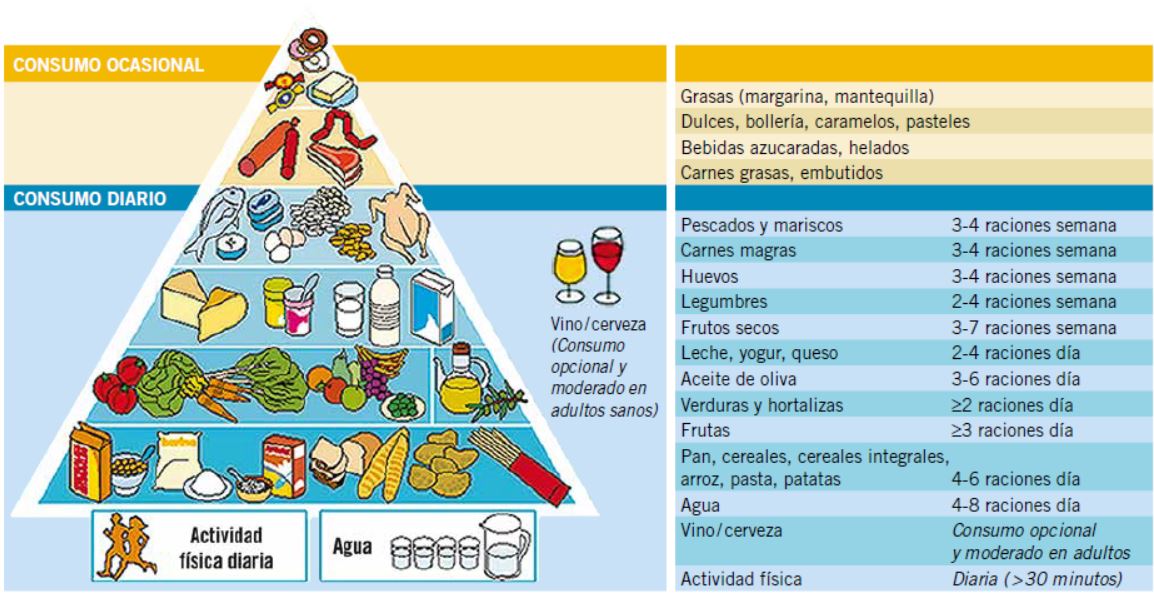
Como otros países, España cuenta con recomendaciones consensuadas por expertos, sobre las orientaciones que deben regir una alimentación saludable; por ejemplo, las publicadas por la Sociedad Española de Nutrición Comunitaria (SENC). Se trata de las Guías Alimentarias para la Población Española, en ellas, se incluye un gráfico en forma de Pirámide Alimentaria (Fig. 1), que permite ver los distintos grupos de alimentos considerados básicos a la vez que muestra las raciones recomendadas de cada grupo(3).
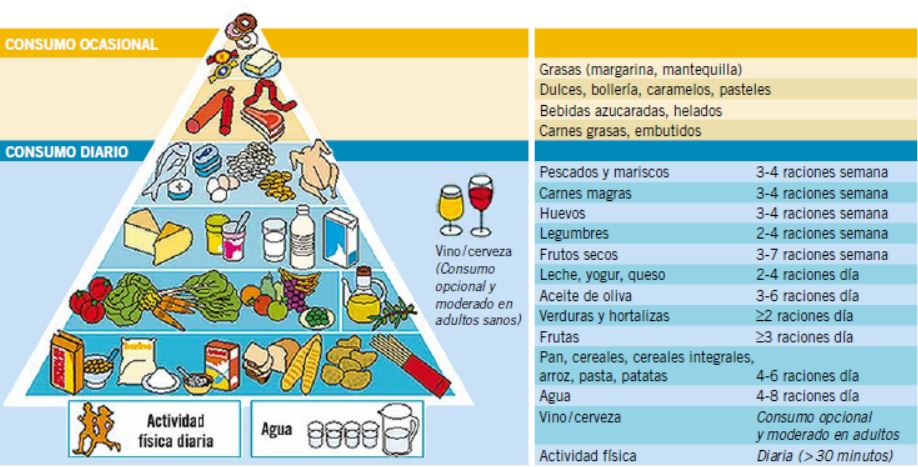
Figura 1. Pirámide de la Alimentación saludable. SENC, 2007.
Alimentaciones no tradicionales
Los padres deben conocer los riesgos y ventajas de la alimentación escogida y recibir información que les ayude a ofrecer a sus hijos una alimentación suficiente.
Por diversos motivos, las personas pueden adoptar un tipo de alimentación distinta a la omnívora. Estas motivaciones pueden ser religiosas (restricciones), ético-filosóficas (no aceptar el sacrificio o captura de animales), ecológicas (impacto ambiental que representa la producción de carne), económicas (la producción vegetal es menos costosa que la animal), fisiológicas (evolución humana de herbívoros a omnívoros), aducidas a problemas de salud o, simplemente, por oposición al sistema establecido.
Históricamente, muchos defensores de actitudes filosóficas y éticas de respeto a la vida y a la paz, han adoptado este tipo de alimentaciones, eminentemente vegetarianas. Entre otros, citamos a Pitágoras, Homero, Séneca, Diógenes, Leonardo da Vinci, Gandhi, Tolstoy, Newton y Victor Hugo.
Una dieta vegetariana bien planificada puede permitir alcanzar la ingesta recomendada para la totalidad de nutrientes, aunque en algunos casos la inclusión de suplementos y alimentos fortificados puede asegurar su aporte(4). Cuando el pediatra se encuentra ante padres con estas inquietudes, debe valorar cuidadosamente el tipo de modalidad que le plantean; es evidente que el riesgo de un desequilibrio nutricional es mayor cuanto menor edad tenga el niño. Por ello, es necesario poder ofrecer a los padres información sobre el tipo de alimentación escogida, sus riesgos y ventajas, e información que les ayude a ofrecer a sus hijos una alimentación suficiente o derivarles a un nutricionista.
Alimentación crudívora
Este tipo de alimentación solo acepta alimentos crudos, aduciendo que las cocciones alteran el valor nutritivo de los alimentos. Va desde los frugívoros, que se alimentan solo de frutos (fruta fresca y seca, aceitunas y frutos grasos), hasta los que amplían su ingesta con hortalizas frutales (tomate, pepino…). Algunos admiten cereales y legumbres (remojados o/y germinados para mejorar parcialmente su digestión), en algunos casos, también incluyen alimentos crudos de origen animal como leche, carne o pescado, y el pan como excepción de alimento cocido.
Alimentación macrobiótica
Patrón dietético propugnado por Georges Oshawa en el marco de la filosofía zen que desea hallar el equilibrio entre fuerzas antagónicas a la vez que complementarias (Yin-Yan).
La aplicación de la alimentación macrobiótica se realiza por fases. En sus inicios, incluye alimentos de origen animal, pero progresivamente se orienta hacia una alimentación vegana con predominio de cereales integrales y limitación del consumo de líquidos. El riesgo de desequilibrios y déficits nutritivos aumenta con el progreso de las restricciones.
Alimentación higienista
Es un sistema que se basa en criterios digestivos y consiste en promover la disociación de determinados alimentos. Por ejemplo, no consumir en la misma comida alimentos proteicos y glucídicos.
Alimentaciones vegetarianas
Las alimentaciones vegetarianas incluyen todo tipo de alimentos de origen vegetal: cereales, legumbres, tubérculos, verduras y hortalizas, frutas, aceites y grasas vegetales y también semillas y frutos secos.
Los alimentos de origen animal y sus derivados son excluidos; pero, en ciertos casos, se aceptan: los productos lácteos, los huevos, la carne de aves o el pescado, de aquí derivan las distintas modalidades vegetarianas que se describen a continuación.
La forma ovolactovegetariana se basa mayoritariamente en alimentos de origen vegetal con inclusión de lácteos y huevos. Es más completa que la lactovegetariana que excluye los huevos y también que la ovovegetariana que excluye los lácteos.
Los vegetarianos estrictos (también llamados veganos, veganistas o vegetalianos) son los que eliminan todo tipo de alimentos de origen animal y que pueden presentar ciertos riesgos nutricionales si no se dispone de información suficiente.
Las últimas crisis alimentarias y el interés de un sector de la población por el seguimiento de unas normas de alimentación saludables, ha fomentado la aparición de una nueva modalidad denominada semivegetariana. Este término abarca un amplio rango de hábitos alimentarios: desde tomar todo tipo de carne de vez en cuando, a consumir huevos, pescados y aves, excluyendo las otras carnes, hasta los que solo aceptan los huevos y el pescado(5).
La tabla I resume las modalidades de vegetarianismo descritas. El presente artículo se centrará en la alimentación vegetariana.

Alimentos
Las pirámides de la alimentación son documentos gráficos que agrupan los alimentos según su semejanza nutricional.
La pirámide para la alimentación vegetariana, al igual que la de alimentación saludable omnívora, agrupa los alimentos según su similitud nutricional, con grupos parecidos, aunque en algunos varían los alimentos que los componen (Fig. 2):

Figura. 2. Pirámide vegetariana. Loma Linda University. School of public Healh.2008. Depatment of Nutrition.
• Cereales: preferentemente integrales.
• Leguminosas: incluye productos derivados (p. ej., tofu).
• Hortalizas: tubérculos, bulbos, raíces y hortalizas de hoja, tallo o fruto.
• Frutas: variadas atendiendo la estacionalidad.
• Semillas oleaginosas y frutos secos.
• Aceites y grasas.
• Lácteos: leches, leches fermentadas de diferentes tipos y quesos.
• Huevos.
Además, el mercado ofrece una amplia gama de productos elaborados para este colectivo sin presencia de alimentos de origen animal.
A continuación, comentamos los contenidos nutricionales de los alimentos básicos por grupos.
Farináceos
Cereales
Los hidratos de carbono son el componente mayoritario de los cereales, en la mayoría de ellos domina el almidón, mientras que en otros como la cebada, la avena o el centeno, predominan los polisacáridos no amiláceos.
Las proteínas son otros componentes no despreciables de su composición. El valor nutritivo de las proteínas de los diversos cereales varía según cada uno de ellos, pero es común para todos los bajos contenidos en lisina si se compara con la proteína patrón, por lo que se las considera de valor biológico limitado. Las proteínas de los cereales se complementan bastante bien con las de origen lácteo, que son mucho más ricas en lisina, así como con las proteínas procedentes de las leguminosas.
Además, los cereales contienen vitaminas del grupo B y sales minerales en cantidades variables, dependiendo de si se considera el grano completo o si este se ha desprovisto de su parte más exterior, lo que da lugar a las harinas refinadas o harinas blancas. Generalmente, las personas que realizan una alimentación vegetariana prefieren consumir cereales completos o alimentos elaborados con harinas integrales, en este caso, el aporte de fibras es mayor, así como el de vitaminas y sales minerales. Los productos integrales tienen una menor digestibilidad y pueden provocar molestias gástricas a los niños y a los adultos poco acostumbrados a su consumo. Los fitatos, que se encuentran en la composición del salvado de los cereales, actúan como inhibidores de la utilización de sales minerales, como el hierro, el cinc y el calcio, acción que se ve disminuida si estos están modificados por la cocción o por fermentación.
Como alimentos de uso común en este subgrupo, se encuentran: el pan, pastas alimenticias, arroz, cereales de desayuno, y entre los elaborados de consumo muy extendido están las galletas y similares.
Legumbres
Son un pilar de la alimentación vegetariana, su composición nutricional está presidida por las proteínas y los hidratos de carbono, sin menospreciar su contenido en sales minerales, vitaminas y fibra. Se consideran una fuente proteica importante. Por su alto contenido en proteínas, se incluyen también en el grupo de alimentos proteicos.
Tubérculos
Por su alto contenido en carbohidratos, los tubérculos también se incluyen en este grupo.
Hortalizas
El contenido en nutrientes de este grupo de alimentos varia de forma considerable según el tipo, pero, en general, destaca la elevada cantidad de agua, que oscila entre el 80 y el 90%, los hidratos de carbono le siguen con un 10-20%, las proteínas y las grasas representan un bajo porcentaje; por todo ello, el contenido energético de las hortalizas frescas es muy bajo. La cantidad de fibras no suele sobrepasar el 3%.
Las hortalizas son una excelente fuente de determinadas vitaminas (folatos, provitamina A, vitamina K, vitamina E, etc.), sobre todo, considerando que esta aportación está asociada a un bajo contenido energético. No obstante, otras vitaminas están prácticamente ausentes en este grupo de alimentos (vitamina D, vitamina B12).
Los alimentos de este grupo son una buena fuente de sales minerales como: potasio, calcio y magnesio. Algunas verduras contienen cantidades relativamente altas de hierro, aunque su biodisponibilidad es baja al igual que la del calcio.
Tubérculos
Deben distinguirse del resto de hortalizas, por el hecho de que su contenido en hidratos de carbono y proteínas es considerablemente mayor al de la mayoría de hortalizas y, por consiguiente, son más energéticos. Tubérculos de uso común en nuestro entorno son: las patatas, los boniatos, pero también, el ñame, y la yuca.
Frutas
Como en el grupo anterior, el componente mayoritario es el agua, a este le siguen los azúcares, los polisacáridos y los ácidos orgánicos, sin apenas presencia de proteínas ni de grasas excepto el aguacate, que contiene una cantidad nada despreciable de grasa, y especialmente de ácidos grasos monoinsaturados. Las vitaminas, las sales minerales y las fibras son parte de las propiedades nutritivas de los alimentos de este grupo, el contenido en vitaminas está distribuido de forma irregular en las distintas especies, por este motivo es importante el consumo variado de las frutas. Las frutas desecadas (pasas, ciruelas…) tienen una cantidad mayor en azúcares, fibras, sales minerales y una cantidad no despreciable de compuestos nitrogenados. Como en el caso de las hortalizas, las vitaminas D y B12 no forman parte de su composición.
Frutos secos y semillas oleaginosas
La fruta seca grasa y las semillas oleaginosas contienen proteínas, además de un considerable porcentaje en grasa, mayoritariamente insaturada. Los ácidos grasos de las almendras, avellanas y pistachos son en su mayoría monoinsaturados, mientras que los de las nueces y los piñones son principalmente poliinsaturados.
En relación a las proteínas, estas son de limitado valor biológico. Aunque son una buena fuente de triptófano y de aminoácidos azufrados, contienen bajos niveles de lisina. Los frutos secos tienen una alta densidad energética que junto con el contenido en vitamina E, sales minerales y fibra, les convierte en un grupo importante para el aporte de nutrientes y energía.
Grasas
Los aceites y las materias grasas son la principal fuente de energía y son el vehículo de las vitaminas liposolubles, de las que podemos destacar la vitamina E en los aceites de semillas o en el aceite de oliva virgen.
Los ácidos grasos de los aceites de semillas son, en su mayoría, poliinsaturados ricos en ácidos grasos de la serie omega-6. El ácido graso predominante del aceite de oliva es el ácido oleico (monoiinsaturado); mientras que, en los aceites tropicales de coco y palma, los ácidos grasos mayoritarios son los ácidos grasos saturados. La mantequilla contiene ácidos grasos saturados, vitamina D y colesterol.
Lácteos
Su contenido en proteínas de alto valor biológico mejorarán el aporte en aminoácidos esenciales sin necesidad de tener en cuenta la complementación proteica; además, contienen una pequeña cantidad de cianocobalamina(B12), vitamina prácticamente ausente de los alimentos de origen vegetal y principal déficit en el vegetarianismo estricto. El calciode los productos lácteos se diferencia, no solo por la cantidad de este mineral, sino también por su buena biodisponibilidad, debida a su forma química y al equilibrio con el fósforo, entre otras buenas condiciones.
La grasa láctea es rica en ácidos grasos saturados. En los casos en que el consumo de alimentos de este grupo sea elevado, puede ser recomendable aconsejar los productos semidesnatados para evitar un excesivo aporte de energía, ácidos grasos saturados y colesterol.
Alimentos de este grupo son: la leche de origen animal y los yogures y demás leches fermentadas, además de los quesos.
Alimentos proteicos
Huevos
Contienen proteínas de alto valor biológico, además de lecitina, fosfolípidos y vitaminas hidrosolubles y liposolubles. El contenido en colesterol no parece resultar un inconveniente, teniendo en cuenta que se trata de un tipo de alimentación donde están ausentes las carnes, los pescados y sus derivados.
Legumbres
Este grupo de alimentos contiene una gran cantidad de proteínas. La calidad de estas, depende de su contenido en aminoácidos esenciales en comparación con la proteína patrón; a los aminoácidos esenciales que resultan insuficientes de esta comparación, se les denomina aminoácidos limitantes.
Las legumbres tienen niveles bajos de triptófano y aminoácidos azufrados, por lo que sus aminoácidos limitantes serán, además del triptófano, la metionina y la cistina. Las legumbres son una buena fuente de lisina(6).
Se puede mejorar el aporte de los aminoácidos, haciendo una combinación adecuada de los distintos grupos de alimentos; por ejemplo, combinando las legumbres, cuyo aminoácido limitante es el triptófano y los aminoácidos azufrados pero que contienen una buena cantidad de lisina, con los cereales, cuyo aminoácido limitante es la lisina y son una buena fuente de triptófano y aminoácidos azufrados.
La calidad de las proteínas también depende de su digestibilidad, que viene determinada por los factores no proteicos como la fibra y los polifenoles, entre otros. A mayor cantidad de fibra, menor es la digestibilidad.
Mediante la fórmula adaptada por la FAO OMS y adoptada por otras organizaciones, se calcula la digestibilidad de una proteína, corregida para el contenido de aminoácidos (PDCAAS); así, la calidad de la proteína de la caseína o la de la clara de huevo tiene un valor 1, mientras que la de las legumbres es de 0,55.
Contenido en aminoácidos
(mg/g proteína) en la
proteína x digestibilidad
PDCASS =
_________________________
Contenido de aminoácidos
en el patrón de la FAO
para niños de 2 a 5 años
En cuanto a los hidratos de carbono, el almidón es el glúcido mayoritario de las legumbres, otros componentes hidrocarbonados son: la celulosa, la hemicelulosa y las pectinas, componentes de las fibras presentes en cantidad importante en estos alimentos. Las grasas son minoritarias en su composición.
Las legumbres ofrecen contenidos interesantes de: ácido fólico, tiamina, niacina, calcio, hierro, cinc, fósforo y magnesio(6).
El hierro de las legumbres, como ocurre con el de los demás alimentos vegetales, es de baja biodisponibilidad. La biodisponibilidad del hierro depende, en parte, de su forma química: el hierro (“hemo” de alta biodisponibilidad), se encuentra solo en músculo y vísceras de animales, mientras que el hierro “no hemo” (con una biodisponibilidad mucho más baja), es el mayoritario en la dieta y se halla presente tanto en alimentos de origen animal como vegetal. Otros factores que afectan la biodisponibilidad del hierro, son la acidez en el medio gástrico, así como la interacción con otros compuestos químicos presentes que en los alimentos pueden aumentar su biodisponibilidad (vitamina C o ácido cítrico) o bien disminuirla (ácidos fítico y oxálico). Además, cuando se ingieren elevadas cantidades de calcio, este compite con el hierro “no-hemo” para su absorción, de manera que puede disminuir su biodisponibilidad. La biosiponibilidad de otros minerales del mismo grupo (minerales de transición como el cinc o el cobre) también se ve afectada por esas interacciones.
Por su contenido en proteínas, también se incluyen en este grupo: los lácteos, los frutos secos y las semillas oleaginosas.
Dulces
Los nutrientes del azúcar y de la miel son, básicamente, azúcares sencillos que aportan energía rápida, la recomendación que se hace sobre el consumo de estos azucares es de no sobrepasar entre el 7-10% de la energía total del día. Su contenido en otros nutrientes es minoritario, especialmente en los refinados. El azúcar moreno y las melazas contiene un poco de fibra y trazas de micronutrientes sin valor destacable en el plano nutritivo.
Alimentos especiales
Alimentos que son consumidos frecuentemente por sus propiedades nutritivas, como condimentos o como sustitutivos de los productos de origen animal. De entre ellos, destacamos:
• Algas: la mayoría de las que se consumen son de origen marino, de bajo contenido calórico, ricas en minerales (Mg, Ca, P, K y I), fibras, proteínas, vitaminas y ácidos grasos esenciales. El contenido vitamínico varía según la época del año y la variedad. Las algas rojas son especialmente ricas en provitamina A, las negras y las verdes en vitamina C. Las negras también lo son en vitamina E. Todas ellas contienen pequeñas cantidades de vitamina B12, si bien algunos autores creen que son análogos inactivos de esta vitamina o bien vitamina activa producida por microorganismos adheridos a ellas, pero que no puede ser considerada como una fuente fiable de esta vitamina. Entre las algas más consumidas, destacan: agar-agar, nori, wakame, hijiki y kombu. Algunas de las que también se consumen son de agua dulce, como en el caso de la espirulina.
• Batidos de soja, arroz, avena: obtenidos a partir de granos remojados, molidos y colados. Usados como sustitutos de la leche, en muchos casos, enriquecidos en calcio.
• Gomasio: condimento obtenido a partir de la mezcla de granos de sésamo tostados y molidos, con sal marina. Existe una variedad mezclada con algas tostadas y pulverizadas, con mayor contenido en minerales.
• Granos germinados: granos sometidos a un proceso germinativo que mejora su digestibilidad.
• Miso: pasta rica en sodio que resulta de la fermentación de granos de soja, con avena o/y arroz, se encuentran diversas variedades según la combinación de los ingredientes. Se usa para la preparación de sopas, como condimento o para untar pan. Su riqueza en ácido glutámico le proporciona el sabor típico de la carne.
• Natto: producto de soja fermentada de manera similar al tempeh. Característico por su textura y sabor peculiar.
• Proteína de soja texturizada: se obtiene de la harina de soja desgrasada. Se comercializa generalmente deshidratada y es muy utilizada por la industria alimentaria.
• Quorn: microproteína que se extrae de un hongo Fusarium venenatum; se consume como ingrediente de algunas recetas culinarias.
• Seitán: preparado proteico a base de gluten de trigo. El valor biológico del gluten del trigo es de 42/100. Permite preparaciones culinarias similares a la carne.
• Tamari: compuesta de soja fermentada, trigo y sal. Se usa como salsa o para condimentar sopas. Tiene un alto contenido en sodio.
• Tahin: Pasta elaborada con semillas de sésamo crudas o tostadas. Usada para untar en pan o para dar sabor a las recetas culinarias.
• Tempeh: producto de soja fermentada a partir de granos enteros de soja. De sabor similar a los champiñones frescos, se suele utilizar como condimento de otros platos.
• Tofu: también llamado “queso de soja”, se obtiene a partir de la coagulación del batido de soja. Son mejor fuente de calcio, los cuajados a los cuales se ha adicionado sales de calcio.
El valor nutricional de algunos de estos alimentos se resume en la tabla II.

Realización de la alimentación vegetariana
Para conseguir una alimentación vegetariana saludable, se recomienda cumplir con las raciones propuestas de cada grupo de alimentos, procurando que sea lo más variada posible y adaptando las cantidades a la necesidades individuales. Para facilitar su confección, en la tabla III se resumen el número de raciones recomendadas por día, así como el peso y la medida casera.

En la tabla IV, se muestran los pesos orientativos de las raciones por grupos de edad.

Análisis crítico de las alimentaciones vegetarianas. Ventajas e inconvenientes
Las dietas vegetarianas ofrecen beneficios de protección de la enfermedad coronaria, debido a su menor contenido en grasa, colesterol, y a su superior contenido en ácido fólico y antioxidantes. Los principales inconvenientes son: el volumen de alimentos que deben consumir para cubrir las necesidades energéticas y la ausencia de determinados nutrientes en alimentos de origen vegetal.
Debido al incremento de las enfermedades crónicas relacionadas en gran parte con la dieta, las recomendaciones actuales de la OMS para la población general, son la disminución del consumo de grasas saturadas y azúcar, y el aumento de fibras, que traducido en alimentos propone la disminución de los alimentos de origen animal y el aumento de los de origen vegetal.
Gibson y colaboradores (2014), describen que existe riesgo de deficiencia de hierro y cinc en niños y adolescentes que practican dietas veganas muy restringidas, por lo que puede ser conveniente recomendar alimentos fortificados, así como estrategias para mejorar la biodisponibilidad(8).
Tanto la Asociación Americana de Dietética (ADA) como la Société Canadienne de Pédiatrie, coinciden en afirmar que una alimentación vegetariana bien equilibrada, puede responder a las necesidades de los niños y de los adolescentes, pero que debe de asegurarse un suficiente aporte energético y vigilar atentamente su desarrollo(9), en este punto, la ADA especifica que es compatible con un buen estado nutricional, especialmente si la dieta es lacto u ovolactovegetariana(4).
En los casos en que la alimentación vegetariana aporte los nutrientes y la energía necesaria para el crecimiento y desarrollo del niño, las ventajas a destacar son: la disminución de las grasas saturadas (siempre que se respeten las raciones recomendadas); el adecuado aporte en ácido fólico –a menudo insuficiente en la alimentación omnívora– y el aporte de fibra.
La Asociación Americana de Dietistas (ADA), resalta que los niños vegetarianos raramente son obesos y, también, que su crecimiento es más lento.
Diversos estudios indican que los vegetarianos presentan tasas inferiores de morbilidad y mortalidad relativas a las diversas enfermedades crónicas en comparación con los no vegetarianos. Las dietas vegetarianas ofrecen beneficios de protección de la enfermedad coronaria, debido a su menor contenido en grasa, colesterol, proteína animal y a su superior contenido en ácido fólico y vitaminas antioxidantes. Los niveles de colesterol total en sangre y de colesterol LDL son, por lo general, inferiores en los vegetarianos. Los inconvenientes de la alimentación vegetariana en niños son: el volumen de alimentos que deben consumir para cubrir sus necesidades energéticas, la menor digestibilidad de los alimentos vegetales –en especial referencia a la digestibilidad de las proteínas– y la falta de nutrientes presentes en alimentos de origen animal, pero ausentes (o casi ausentes) en alimentos de origen vegetal.
Cubrir las necesidades en vitamina B12, vitamina D, calcio, zinc y hierro, en los casos de vegetarianismo estricto, puede resultar difícil, así como asegurar el aporte de ácidos grasos esenciales. En algunos casos, se recomienda la suplementación y el consumo de alimentos enriquecidos, como se ha mencionado anteriormente.
Propuestas para mejorar los aportes nutricionales en niños y adolescentes vegetarianos
Los padres deben disponer de la información suficiente para seguir las directrices de la pirámide alimentaria y cumplir con las recomendaciones para mejorar el aprovechamiento de los nutrientes.
Para evitar el déficit energético y/o nutricional, los padres deben conocer y seguir las directrices de la pirámide alimentaria, cumplir con las recomendaciones sobre complementación proteica, aporte de energía, ácidos grasos esenciales, vitamina B12, vitamina D, calcio, zinc y las propuestas para mejorar la utilización del hierro. Para evitar déficit de vitamina B12 y vitamina D en la leche materna de madres veganas, puede ser aconsejable que estas madres las ingieran como suplementos vitamínicos.
Recomendaciones para el aporte de energía
Conviene asegurar un aporte diario de grasas de aliño y cocción, velar por la presencia de cereales o sus derivados en todas las comidas, recomendar el consumo frecuente de fruta seca oleaginosa, pero moderar el consumo de alimentos con mayor contenido en azúcar (zumos, mermeladas, miel, azúcar, chocolate…).
Recomendaciones para la complementación proteica
En el caso de no incluir ningún alimento de origen animal (lácteos o huevos), la mezcla de cereales con legumbres o con fruta seca oleaginosa debería de estar presente en los menús de forma habitual. Las investigaciones sugieren que no es necesario consumir proteínas complementarias en una misma comida y que el consumo de diversas fuentes de aminoácidos durante el día debe asegurar una retención y utilización adecuada del nitrógeno en las personas sanas. Se recomienda un consumo superior de proteínas a los vegetarianos en relación a los no vegetarianos, de entre un 30-35% respecto al valor calórico total para los niños de hasta los 2 años, entre un 20-30% entre los 2 a 6 años, y un 10-15% después de los 6 años, en especial los niños veganos(9), y un consumo variado de alimentos para asegurar la adecuada complementación de las distintas proteínas vegetales. Las fuentes principales de proteínas en la alimentación vegana son: las legumbres, los cereales, los frutos secos y las semillas.
Recomendaciones para el aporte en vitamina B12
La manera más segura es la inclusión de alimentos de origen animal. En ausencia o en un consumo muy bajo de estos, es necesaria la suplementación de esta vitamina o/y el consumo de alimentos enriquecidos, como se ha mencionado anteriormente.
Es posible que la leche materna de madres veganas sea pobre en esta vitamina, por lo que pueden ser necesarios suplementos a sus lactantes(9).
Recomendaciones en relación al aporte de calcio
Los lácteos serán la primera fuente de elección, no solo por la cantidad sino también por la alta biodisponibilidad del mineral en estos alimentos. En los casos de omisión total de los alimentos de este grupo, debe recurrirse a los productos enriquecidos en este mineral como derivados de la soja, cereales, y zumos. Los frutos secos grasos son también portadores de cantidades no despreciables de calcio. Las hortalizas de hoja verde con bajo contenido en oxalatos como la col verde, son también una fuente de calcio para niños de mayor edad. La ingesta recomendada de calcio para niños de 1 a 10 años es de 800 mg/día y de 11 a 24 años es de 1.200 mg/día, cantidades que es importante respetar. Los niños y adolescentes veganos pueden necesitar suplementación para alcanzar las ingestas recomendadas.
Propuesta para mejorar la utilización del hierro
A partir de los 6 meses, deben introducirse alimentos ricos en hierro o alimentos enriquecidos en este mineral.
Diversos estudios realizados con niños preescolares y escolares veganos, indican que no hay casos documentados de anemia. Si bien, su necesidad en este mineral es de 1,8 veces en relación a los no vegetarianos, debido a la diferente biodisponibilidad(9).
Para mejorar la biodisponibilidad del hierro de los alimentos de origen vegetal se recomienda la inclusión de un alimento rico en vitamina C en la misma comida, ya que la vitamina C incrementa la solubilidad del hierro “no-hemo” (y, por consiguiente, su biodisponibilidad). Altas concentraciones de ácidos fítico u oxálico, cantidades altas de fibra y elevadas ingestas de calcio, también interfieren negativamente en la absorción de este mineral.
Propuesta para mejorar el aporte de zinc
Debido a la baja biodisponibilidad del zinc de los alimentos de origen vegetal, las recomendaciones de consumo para los vegetarianos y, en especial, para los veganos deberían de ser un 50% superior a las de los no veganos. Fuentes de este mineral son (legumbres secas, fruta oleaginosa y cereales completos). La fermentación de los alimentos permite disminuir las concentraciones de posibles sustancias interferentes(4,8,9,11,12).
Propuesta para mejorar el aporte de vitamina D
Para los veganos, el aporte de esta vitamina está supeditada al consumo de alimentos enriquecidos y a la exposición solar, en el caso de que estos sean insuficientes, es muy posible que sea necesaria la prescripción de suplementos.
Propuesta para mejorar el aporte de ácidos grasos esenciales
Los veganos pueden fácilmente consumir menos grasas que los omnívoros y los ovo-lactovegetarianos, siendo especialmente bajas las ingestas de docosahexaenoico (DHA) y de eicosapentanoico (EPA) y, en cambio, altas las ingestas de ácidos grasos omega-6, los cuales pueden inhibir la conversión del ácido linolénico, principal precursor del DHA y del EPA, por lo que se recomienda el consumo de nueces, aceite de linaza y aceite de colza ricos en el precursor.
Función del pediatra de Atención Primaria
El pediatra debe velar para que los aportes nutritivos sean adecuados, teniendo en cuenta la edad de los niños, para que estos mantengan un óptimo estado de salud y desarrollo, además de prevenir trastornos alimentarios que se pueden traducir en desequilibrios nutricionales y con ello favorecer la aparición posterior de: obesidad, hipercolesterolemia, diabetes tipo 2, caries y malnutrición en general. Dichos trastornos y patologías crecen actualmente a un ritmo no deseable que generan una mala calidad de vida y un coste socio-económico y sanitario muy elevado. También, debe prevenir aportes energéticos insuficientes y desequilibrios nutricionales para evitar déficits nutricionales.
Promocionar el seguimiento de las recomendaciones de la pirámide vegetariana, junto con una buena información, puede ser útil para conseguir una alimentación saludable.
Cualquier tipo de alimentación puede ser saludable siempre que cumpla con los requisitos nutricionales. Dicho de otra forma: que sea suficiente en energía y nutrientes(13).
Bibliografía
Los asteriscos reflejan el interés del artículo a juicio del autor.
1. Sabaté J, Soret S. Sustainability of plant-based diets: back to the future. Am J Clin Nutr. 2014; 100 (suppl): 476S-82S. American Society for Nutrition.
2. Cervera P, Alimentació Maternoinfantil. 2a ed. Barcelona: Masson; 2000.
3.*** Guías alimentarias para la población española: recomendaciones para una dieta saludable.Madrid: Sociedad Española de Nutrición Comunitaria; 2007.
4.*** Craig WJ, Mangels AR; American Dietetic Association. Position of the American Dietetic Association: vegetarian diets. J Am Diet Assoc. 2009; 109(7): 1266-82.
5.** Puiggròs C, et al. Dieta vegetariana En: Salas J, et al. Nutrición y dietética clínica. Barcelona: Masson; 2014. 453-63.
6.*** Messina V, Mangels AR. Dietician’s Guide to Vegetarian diets: Issues and Applications. 2ed. Jines & Bartlett Publishers; 2004.
7.** Astiasarán I, Martínez JA. Alimentos: composición y propiedades. Madrid: McGraw-Hill Interamericana; 2000.
8.** Gibson RS, et al. Is Iron and zinc Nutrition a concern for vegetarian infants and Young children in industrialized countries? Am J Clin Nutr. 2014; 100 (suppl): 459S-68S. American Society for Nutrition.
9.*** Amit M. Les régimes végétariens chez les enfants et les adolescents. Comité de la pédiatrie communautaire. Societé canadienne de pédiatrie. Paediatr Child Health. 2014; 15(5): 309-14.
10.*** Messina V, Mangels AR. Considerations in planning vegan diets: children. J Am Diet Assoc. 2001; 101(6): 661-9.
11.* Rigolfas R, Padró L y Cervera P. Educar en la alimentación y la nutrición. Tibidabo Ediciones. Barcelona 2010.
12.*** Mangels AR, Messina V. Considerations in planning vegan diets: infants. J Am Diet Assoc. 2001; 101(6): 670-7.
13.*** Basulto J. Dietas vegetarianas en momentos de riesgo del ciclo vital. Grupo de Revisión, Estudio y Posicionamiento de la Asociación Española de Dietistas-Nutricionistas GREP-AEDN. 2007.
14. CESNID. Tablas de composición de alimentos. Barcelona: Universidad de Barcelona; 2002.
15. USDA National Nutrient Database for Standard Reference, Release 15 (August 2002): http://www.nal.usda.gov.
16. Absolone, J. L’alimentacion vegetarienne. Institut Paul Lambin, 1995.
Bibliografía recomendada
- Bras J, de la Flor JE, Masvidal RMª. Pediatría en Atención Primaria. 3ª ed. Barcelona: Springer-Verlag Ibérica; cop. 2011.
Publicación redactada por pediatras de Atención Primaria, que trabajan con el entorno familiar cotidiano donde se desarrolla el niño, ayudando en los problemas que plantea su crecimiento y desarrollo, el rendimiento escolar, la socialización con los compañeros, el acoplamiento a la sociedad y, el seguimiento y recuperación de posibles secuelas patológicas y minusvalías.
- Pich M. Niños vegetarianos En: Sasot J, Moraga FA. Avances en psicopediatría. Barcelona: Prous Science; 2001. p. 37-48.
Libro que tiene la educación como el punto de partida para la prevención de la mayoría de los problemas de salud, tanto físicos como psicosociales, que pueden transformar las etapas del desarrollo y de la madurez en un camino difícil e inseguro con consecuencias que afectarán negativamente la conducta, la salud y la propia vida.
- Centre d’Ensenyament Superior de Nutrició i Dietètica. L’alimentació infantil: nens i nenes de 3 a 12 anys. Barcelona: Pòrtic; 2001.
Publicación que ofrece consejos, pautas y recomendaciones alimentarias para la adecuada alimentación infantil. Además, contiene ciento cincuenta recetas con el comentario dietético y el análisis de la cobertura de raciones alimentarias.
- Les algues: le légume de la mer. Nutri-doc. 2002; (38): 1.
Artículo que da a conocer, que más de nueve millones de toneladas de algas son transformadas cada año en el mundo, de las cuales el 75% son utilizadas como verduras. Tradicionalmente, estas algas son más utilizadas en el sureste asiático que en Occidente donde todavía tienen pocos adeptos.
- Lecerf JM. Manger autrement. 3è éd. Paris: Institut Pasteur de Lille; 1991.
Obra sencilla, divertida y clara, donde el autor propone toda clase de recetas y alternativas culinarias para alimentarse de manera saludable y equilibrada.
- UVE. http://www.unionvegetariana.org/.
Web de la Unión vegetariana española, que cuenta con abundante y riguroso material de interés para la realización práctica de la alimentación vegetariana, así como respuestas fundamentadas científicamente sobre muchos aspectos que plantean dudas a los practicantes de estas dietas.

| Caso clínico |
|
Adolescente de 16 años que acude a consulta derivada por el ginecólogo debido a una amenorrea de tres meses, de la analítica realizada destacan unos hematíes de 3.800.000/ml, un hematocrito del 34%, un VCM de 78 fl y una ferritina de 12 ng/L, el resto de los valores están dentro de la normalidad. La madre explica que desde hace aproximadamente 10 meses, su hija ha decidido seguir una alimentación vegetariana, ella misma reconoce que es afín a este tipo de alimentación, pero que su hija la sigue de forma estricta. El interrogatorio alimentario realizado demuestra que la muchacha sigue una alimentación vegetariana estricta. Pero no demuestra demasiados conocimientos sobre alimentación y no parece haberse informado sobre el veganismo. Habitualmente, hace todas las comidas en su casa excepto la del mediodía que la realiza en el centro escolar donde estudia, allí acostumbra a comer el primer plato del menú: verdura y patata o pasta con salsa de tomate o ensalada, además, come pan y fruta. En casa, la alimentación se compone esencialmente de pasta, arroz, hortalizas, patatas, pan, fruta, pocos frutos secos y productos derivados de la soja, como batido de soja, tofu, yogur de soja y miso. También consume seitán frecuentemente. Mantiene la misma actividad física desde que empezó la ESO. El IMC es de 22 y su peso no ha variado desde el cambio de su alimentación.
|