 |
| Temas de FC |
F. Santos Rodríguez
Universidad de Oviedo. Hospital Universitario Central de Asturias, Oviedo
| Resumen
Las tubulopatías constituyen un grupo de enfermedades heterogéneas, raras y complejas. Este artículo se centra en aspectos comunes o compartidos de las tubulopatías de base genética o primarias, que son las que se presentan típicamente en la edad pediátrica. Se discuten las alteraciones que estas entidades causan en diferentes sistemas metabólicos del organismo: principios inmediatos, equilibrio ácido-base e hidroelectrolítico, metabolismo mineral y homeostasis del ácido úrico. Se delimita un perfil clínico y bioquímico común que debe de llevar a la sospecha diagnóstica. Se indican, asimismo, las bases del diagnóstico genético, del tratamiento y del pronóstico y se señalan de forma resumida las características principales de algunas tubulopatías representativas. El artículo proporciona referencias actualizadas sobre revisiones y guías clínicas recientemente publicadas que permitirán profundizar en el estudio y conocimiento de estas enfermedades. |
| Abstract
Tubulopathies are a group of heterogeneous, rare and complex diseases. This article focuses on common aspects of primary tubulopathies of genetic basis, which are those typically presenting in the pediatric age. The alterations caused by these diseases in the organism’s metabolic systems are discussed: glucose and amino acid metabolisms, acid-base balance, water and electrolyte equilibrium, mineral metabolism and uric acid homeostasis. A common clinical and biochemical profile to suspect the diagnosis is established, as well as the basis of genetic diagnosis, treatment, and outcome. The main features of some representative tubulopathies are summarized. The article provides updated references on reviews and clinical guides recently published to facilitate a further in-depth study and knowledge of these diseases. |
Palabras clave: Tubulopatías; Función tubular; Enfermedades genéticas; Nefropatías primarias.
Key words: Tubulopathies; Tubular function; Genetic diseases; Primary nephropathies.
Pediatr Integral 2022; XXVI (8): 482 – 491
OBJETIVOS
• Las bases del diagnóstico y los fundamentos del tratamiento de las enfermedades tubulares de base genética.
• El espectro de alteraciones clínicas, bioquímicas y de imagen que permiten establecer la sospecha diagnóstica de tubulopatía en un paciente pediátrico.
• Las características más importantes de algunas tubulopatías representativas.
• Recursos bibliográficos y de Internet actualizados para profundizar en el estudio de las tubulopatías primarias más significativas.
Tubulopatías
Introducción
Las tubulopatías primarias son enfermedades raras, pero típicamente pediátricas. Es preciso conocerlas para poder detectarlas precozmente, tratarlas y minimizar el riesgo de complicaciones.
El término “tubulopatías” incluye un grupo de enfermedades en las que la afectación de la función tubular renal es predominante sobre la disfunción glomerular y ocurre con anterioridad a esta, que, sin embargo, puede surgir y adquirir relevancia clínica en la evolución a largo plazo.
Las tubulopatías pueden ser primarias, causadas por un defecto genético de una proteína que interviene en las funciones fisiológicas del túbulo renal, y adquiridas o secundarias, en las que la función tubular se altera como consecuencia de la acción de un tóxico, fármaco y/o enfermedad sistémica o de origen extrarrenal. Este artículo se centra sobre las tubulopatías primarias, que son las típicamente pediátricas, ya que las tubulopatías secundarias son más propias de la edad adulta.
Las tubulopatías primarias son enfermedades raras o ultrarraras. Su diagnóstico, frecuentemente complicado, y su tratamiento corresponden habitualmente a unidades especializadas de nefrología pediátrica, pero el pediatra debe de conocerlas, al menos, por las siguientes razones:
• Son entidades característicamente pediátricas, muy poco conocidas en general por los especialistas de adultos.
• Suelen debutar en la edad infantil.
• Su precoz identificación y tratamiento adecuado son de enorme importancia para la salud del niño y para prevenir la aparición de complicaciones a largo plazo.
• Existe posibilidad de establecer un consejo genético que delimite la herencia de la enfermedad, el riesgo de padecerla en futuros hermanos y la opción de detección en la vida prenatal o en las primeras semanas o meses de vida postnatal.
Además de estas razones de impacto clínico directo, el estudio de las tubulopatías y su reflexión sobre sus manifestaciones y base molecular, representan para el pediatra interesado una oportunidad inigualable para conocer la fisiología renal y la participación de los riñones en la homeostasis de diversos sistemas metabólicos del organismo.
Este artículo no pretende describir los diferentes tipos de tubulopatías primarias, sino proporcionar la información necesaria para que el pediatra sepa sospecharlas clínicamente y enfocar su diagnóstico, resaltar aquellas entidades a cuyo conocimiento grupos españoles han hecho aportaciones relevantes, así como indicar recursos de apoyo bibliográfico o por Internet para la búsqueda de información adicional más completa.
Bases fisiopatológicas
Los túbulos renales determinan la composición de la orina y son esenciales para el mantenimiento del medio interno, regulando diferentes sistemas metabólicos.
El túbulo renal a lo largo de sus diferentes segmentos modifica el filtrado glomerular, mediante procesos de reabsorción y secreción, y determina así la composición final de la orina que se elimina. Estos procesos requieren la integridad anatómica de la nefrona y del intersticio renal, la normal estructura de las proteínas responsables de los mecanismos de transferencia de diversas moléculas a través de las células tubulares y de las uniones paracelulares y la disponibilidad de energía necesaria para la actividad de los sistemas de transporte activo. En la regulación de estos procesos intervienen factores físicos y químicos, acciones autocrinas y paracrinas y diversos sistemas hormonales, como: renina – aldosterona, hormona antidiurética y eje paratohormona (PTH) – vitamina D – factor de crecimiento de fibroblastos 23 (FGF23). Numerosas proteínas, bien sean transportadoras, receptoras, canales o enzimas, entre otras, están implicadas en el metabolismo renal de una sustancia, por lo que diversos defectos moleculares pueden conducir a similares alteraciones clínicas o bioquímicas. Por otra parte, existen fenómenos de maduración y de compensación en la nefrona que pueden explicar la cronología de aparición o relevancia de las manifestaciones clínicas durante la infancia. Es de notar, asimismo, que el riñón es un órgano endocrino que no solo produce eritropoyetina, sino que las células del túbulo proximal sintetizan de forma regulada 1,25 dihidroxivitamina D [1,25(OH)2D] o calcitriol, metabolito activo de la vitamina D.
El túbulo renal juega un papel esencial en la homeostasis del organismo, pudiendo distinguirse, desde un punto de vista didáctico, su participación en los siguientes sistemas metabólicos:
• Metabolismo de los principios inmediatos.
• Equilibrio ácido-base.
• Equilibrio hidroelectrolítico.
• Metabolismo mineral: calcio – fósforo – magnesio.
• Metabolismo del ácido úrico.
Es útil recordar con fines clínicos que en el túbulo proximal tiene lugar el grueso de los procesos de reabsorción, que requieren alto consumo de energía por la célula tubular, mientras que en la nefrona distal tienen lugar procesos de regulación “fina” y compensatoria.
Manifestaciones clínicas
Aunque las tubulopatías son entidades distintas y heterogéneas, puede reconocerse un espectro común de síntomas y signos clínicos útil para establecer un diagnóstico de sospecha.
Dependerán de la enfermedad en concreto, pero puede reconocerse un perfil clínico general, con presentación y predominio de unas u otras manifestaciones según el sistema metabólico más afectado. Su conocimiento será de utilidad para que el pediatra establezca la sospecha diagnóstica de tubulopatía. La tabla I muestra índices urinarios de ayuda para el diagnóstico(1-3).

Metabolismo de los principios inmediatos
La disminución de la reabsorción tubular de aminoácidos causa hiperaminoaciduria, específica o generalizada según el defecto molecular subyacente. La pérdida urinaria de aminoácidos puede no producir cambios significativos en los niveles circulantes y depender la clínica únicamente de la acumulación y posible precipitación del aminoácido en la orina, como en el caso de la cistinuria. Una hiperaminoaciduria asintomática puede traducir un defecto de maduración renal o ser una manifestación precoz de una tubulopatía proximal más compleja y/o de una enfermedad mitocondrial.
La glucosuria renal, es decir, la que sucede sin hiperglucemia por defecto en la reabsorción proximal de glucosa, no provoca síntomas. Es importante conocer esta entidad para evitar un diagnóstico erróneo de diabetes mellitus. La glucosuria transitoria cuando, por alto aporte dietético de hidratos de carbono, la glucemia excede el dintel renal fisiológico de glucosa no representa ninguna anomalía.
Equilibrio ácido-base
La acidosis metabólica hiperclorémica o con “anion gap” normal es una manifestación bioquímica característica de la acidosis de origen tubular, si bien hay que tener presente que la causa más frecuente en Pediatría de acidosis metabólica hiperclorémica transitoria son las alteraciones gastrointestinales agudas, como la diarrea, que conducen a la pérdida digestiva de bicarbonato.
Las tubulopatías que cursan con pérdida de sal, depleción de potasio, contracción del volumen vascular e hiperaldosteronismo secundario causan alcalosis metabólica hipopotasémica.
Equilibrio hidroelectrolítico
Las pérdidas excesivas de agua, electrolitos y otros elementos con efecto osmótico ocasionan poliuria y defecto de la capacidad de concentración urinaria, lo que conlleva un alto riesgo de episodios de deshidratación con alteraciones en las concentraciones séricas de electrolitos. Esta situación incide particularmente en niños lactantes o de muy corta edad con dificultad al acceso espontáneo de líquidos, dieta láctea y mayor proporción de volumen extracelular.
Metabolismo mineral: calcio – fósforo – magnesio
En este ámbito, la hipofosforemia por aumento de la eliminación urinaria de fosfato junto con el déficit de síntesis tubular de 1,25(OH)2D explican la aparición de raquitismo vitamina D resistente. La hipercalciuria no ocasiona hipocalcemia, pero sí supone un factor de riesgo para la formación de cálculos urinarios y nefrocalcinosis, sobre todo si se acompaña de hipocitraturia. Asimismo, la hipomagnesemia por pérdida elevada de magnesio en orina es causa de convulsiones, miopatía y alteraciones en la secreción de PTH.
Metabolismo del ácido úrico
La hipouricemia marcada, incluso por debajo de 1 mg/dl, es manifestación típica de algunas tubulopatías proximales con hiperuricosuria que confiere riesgo litógeno.
Las manifestaciones anteriores concurren, en mayor o menor grado, alterando el normal desarrollo y crecimiento del niño, manifestación mayor y muy frecuente de las tubulopatías. Así pues, el pediatra debe de pensar en la posibilidad de tubulopatía primaria subyacente en un lactante, en algunas tubulopatías con mayor prevalencia de bajo peso neonatal, o niño en su infancia temprana con defecto de medro y afectación de la curva estato-ponderal, que presenta episodios de deshidratación con anomalías electrolíticas y del equilibrio ácido base que:
• Se desencadenan con inusual facilidad por cuadros aparentemente banales de rechazo de tomas, vómitos o diarrea.
• Tienen carácter recurrente.
• Tendencia a prolongarse en el tiempo a pesar de las medidas terapéuticas. Asimismo, el hallazgo de raquitismo no carencial, nefrocalcinosis medular o urolitiasis no causada por infecciones urinarias obliga a descartar una enfermedad tubular renal.
Bases genéticas
La identificación del defecto genético responsable es importante para confirmar el diagnóstico clínico, para asesorar a la familia sobre la herencia de la enfermedad y para avanzar en el conocimiento de la enfermedad.
Las tubulopatías primarias son enfermedades de origen genético, la gran mayoría ocasionadas por mutaciones o alteraciones génicas que causan defectos de función de la proteína codificada por el gen(4). En muy pocas entidades existe como base una ganancia de función, el síndrome de Liddle, por ejemplo. En general, son enfermedades de herencia autosómica recesiva (AR), si bien hay importantes tubulopatías ligadas al cromosoma X y en otras la herencia es autosómica dominante (AD). Estas últimas suelen debutar más tarde y tienen clínica menos grave. La historia familiar debe de realizarse en detalle y puede ser contributiva, sin embargo, también puede ser negativa en caso de mutaciones “de novo”.
El defecto puede afectar a genes que se expresan en otros epitelios o tejidos, además del túbulo renal, dando lugar a manifestaciones asociadas extrarrenales, por ejemplo, hipoacusia presente en varias tubulopatías primarias.
La identificación de la alteración genética causal no es requisito indispensable para el diagnóstico de la tubulopatía; ya que este puede hacerse en función de los hallazgos clínicos, de laboratorio y radiológicos. No obstante, debe de procurarse la confirmación del defecto genético-molecular: por la posibilidad de errores diagnósticos, por solapamientos en el fenotipo o por fenotipos no claros, para plantear el diagnóstico prenatal en futuros hermanos y para establecer un consejo genético adecuado. Un estudio genético negativo puede explicarse por la resolución insuficiente de las técnicas disponibles de secuenciación y obliga a revisar el diagnóstico clínico y al análisis, si es posible de otros genes candidatos. La detección de variantes no definitivamente patogénicas no es infrecuente, suscita incertidumbres en el diagnóstico y requiere estudiar a los padres y hermanos. Sería deseable en este caso la realización de estudios complementarios que averigüen si la variante génica causa alteraciones en la función de la proteína, pero esta posibilidad no suele estar al alcance de laboratorios clínicos y queda reservada al ámbito de la investigación. En cualquier caso, la interpretación de los hallazgos del estudio genético exige una estrecha relación para el diagnóstico entre los profesionales del laboratorio que lo han realizado y los clínicos que siguen al paciente.
Con independencia del valor diagnóstico del estudio genético, debe de insistirse en la necesidad de su realización, ya que supone una contribución importante al conocimiento de la enfermedad y de los mecanismos fisiopatológicos causantes de las manifestaciones clínicas. Además de que ha descendido notablemente el coste de los estudios de secuenciación en laboratorios privados, existen numerosas plataformas por Internet, a través de las que puede realizarse un estudio genético gratuito. A este respecto, hay que mencionar RenalTube (www.renaltube.com), un portal de ámbito multicéntrico internacional desarrollado en España, que está disponible para ayudar a los pediatras en el estudio clínico-genético de las tubulopatías primarias.
Así pues, al perfil clínico del niño con tubulopatía primaria, anteriormente descrito, debe de añadirse la posibilidad de identificar consanguinidad o un patrón de herencia ligado al sexo en una historia familiar bien detallada. El hallazgo de una mutación causal puede contribuir a delimitar o predecir las manifestaciones clínicas y el pronóstico de la enfermedad, si bien hay que resaltar que es frecuente que no exista una clara correlación genotipo-fenotipo.
Bases terapéuticas
La dieta, los suplementos de agua, electrolitos o minerales, las medidas farmacológicas dirigidas a disminuir la pérdida hidroelectrolítica, la corrección de déficits hormonales y los medicamentos específicos para contrarrestar el defecto molecular subyacente son las bases del tratamiento.
Aunque el tratamiento de las tubulopatías será específico para cada una de ellas, es importante tener presente algunas consideraciones comunes a muchas de ellas que se indican esquemáticamente a continuación.
Dieta
Acceso libre al agua y a la sal en la dieta. Esencial en aquellas tubulopatías que cursan con poliuria y pérdida de electrolitos. En los primeros meses de vida, el niño no puede autorregular su alimentación y es preciso prestar especial atención a no introducir restricciones dietéticas innecesarias y potencialmente dañinas.
Suplementos
En principio, las pérdidas en la orina derivadas del defecto en la reabsorción tubular deben de compensarse con suplementos de la sustancia correspondiente. Sin embargo, la reposición total de las pérdidas, particularmente en las tubulopatías proximales, es frecuentemente inalcanzable, ya que cuanto más se administre más se pierde, y perseguir la normalización continuada de la concentración sanguínea de la sustancia afecta puede conducir a toxicidad y no ser un objetivo terapéutico deseable.
Reducción del filtrado glomerular
En relación con los dos puntos anteriores, la administración de fármacos dirigidos a reducir la tasa de filtración glomerular (GFR) puede disminuir la carga hidrosalina que llega al túbulo y disminuir la poliuria y la fuga de electrolitos. Con esta finalidad, se han empleado indometacina o inhibidores de la ciclooxigenasa 2 en tubulopatías proximales y en nefropatías pierde sal(5).
Prevención de urolitiasis y nefrocalcinosis
Aminoración del riesgo litógeno mediante aporte alto de líquidos para diluir la orina y disminuir la concentración urinaria de las sustancias que precipitan. Si no es una nefropatía pierde sal, la disminución del aporte de sal contribuirá a este objetivo, por ejemplo, en el caso de la cistinuria. Administración de citrato como inhibidor de la precipitación de sales de calcio.
Sustitución hormonal
La disrupción de la función endocrina del riñón puede requerir la administración de derivados 1-hidroxilados de vitamina D o de mineralocorticoides en algunas tubulopatías concretas. De la misma manera, el tratamiento con hormona de crecimiento (GH) ha mostrado su utilidad para mejorar el crecimiento en ciertas tubulopatías(8), aunque no sea una indicación incluida entre las oficialmente aprobadas por el Ministerio de Sanidad español.
Tratamientos específicos dirigidos a la base patogénica de la enfermedad
Disponibles en los últimos años para algunas tubulopatías graves mediante medicamentos que contrarrestan el defecto molecular subyacente, anticuerpos monoclonales y terapias génicas o con trasplante de células madre que comienzan a ofrecer resultados prometedores.
Prevención y tratamiento de complicaciones a largo plazo y extrarrenales
Aunque las tubulopatías se caracterizan porque la afectación tubular es predominante sobre la glomerular, debe de tenerse en cuenta en el seguimiento de estos pacientes que la insuficiencia renal por reducción del GFR se presenta en muchas tubulopatías en el seguimiento a largo plazo y deben de tomarse las medidas orientadas a prevenir su aparición y enlentecer su progresión. A este respecto, hay que señalar que la proteinuria de bajo peso molecular de las tubulopatías proximales no responde al tratamiento con inhibidores del sistema renina-angiotensina o bloqueantes de los receptores de angiotensina. Por otra parte, ya se ha mencionado la posibilidad de manifestaciones extrarrenales que pueden requerir tratamiento específico.
Pronóstico
La evolución es variable según la tubulopatía, la precocidad del diagnóstico y tratamiento y la gravedad de las manifestaciones extrarrenales.
Variable según la tubulopatía de que se trate. De forma general, puede afirmarse que dependerá de:
• Precocidad en el diagnóstico y en la instauración de un tratamiento correcto que corrija, en la medida de lo posible: las alteraciones existentes, prevenga la aparición de complicaciones en los episodios agudos de descompensación de la enfermedad y evite los efectos tóxicos farmacológicos.
• Gravedad de las manifestaciones extrarrenales asociadas a la tubulopatía o derivadas de la misma.
• Disminución progresiva de la GFR producida por diversos mecanismos no bien conocidos, como lesiones inflamatorias crónicas del intersticio renal o afectación de la permeabilidad de la barrera de filtración glomerular.
Tubulopatías según el sistema metabólico predominantemente afecto. Entidades ilustrativas
Metabolismo de los principios inmediatos
El defecto molecular radica en transportadores específicos del túbulo contorneado proximal.
Glucosuria renal
Enfermedad de herencia AR/AD (OMIM # 233100) causada por defectos de función del gen SLC5A2, que codifica el transportador SGLT2. Los pacientes tienen glucosuria por disminución de la reabsorción tubular de glucosa sin hiperglucemia ni otros signos de disfunción tubular. Es una entidad benigna que no requiere tratamiento.
Cistinuria
Causa importante y tratable de urolitiasis metabólica en niños.
Enfermedad de transmisión autosómica, con un patrón complejo, no siempre AR (OMIM # 220100), caracterizada por defecto en el transporte de cistina y aminoácidos dibásicos (lisina, arginina y ornitina) en el túbulo renal proximal y en el tracto gastrointestinal. Causada por defectos primarios de función en los genes SLC3A1 y/o SLC7A9. La elevada concentración de cistina en la luz tubular y su baja solubilidad en pH no alcalino conducen a la formación de cálculos urinarios recurrentes, con riesgo de uropatía obstructiva, infecciones urinarias y, más raramente, fallo renal(6). La pérdida urinaria de lisina, ornitina y arginina no da síntomas, ya que estos aminoácidos son muy solubles. Sin embargo, la cistina tiene riesgo de precipitar cuando su concentración urinaria es superior a 250 mg/l (1 mmol/l) y el pH es ≤ 7, formando cristales hexagonales, planos y transparentes que originan cálculos radiopacos que pueden ser coraliformes. Los valores normales del cociente urinario cistina/creatinina están en torno a 20 mg/g, aumentando 10 veces o más en pacientes homocigotos y 3-4 veces en heterocigotos. Las medidas terapéuticas van dirigidas a disminuir la concentración urinaria de cistina, mediante aporte alto de líquidos y restricción dietética de sal, y a aumentar su solubilidad, mediante alcalinización de la orina y administración de tiopronina, que forma un compuesto mucho más soluble con la cistina, si las medidas previas no son suficientemente eficaces.
Equilibrio ácido-base
Las tubulopatías más representativas en este apartado son las que se engloban bajo el término “acidosis tubular renal” (ATR), caracterizadas por acidosis metabólica hiperclorémica y que ocurren como consecuencia de una disminución de la reabsorción de HCO3– y/o de la excreción urinaria de H+. Según criterios clínicos, fisiopatológicos, y moleculares, existen 4 tipos de ATR, numerados por orden cronológico de descripción: ATR distal o tipo 1, ATR proximal o tipo 2, ATR tipo 3 (una forma mixta de tipo 1 y 2) y ATR tipo 4 o hiperpotasémica(7).
La ATR proximal ocurre en el contexto de tubulopatías proximales complejas, siendo excepcional como enfermedad aislada. La fuga masiva de bicarbonato requiere altas dosis de álcali para controlar la acidosis. La ATR tipo 4 forma parte de entidades que cursan con baja eliminación de amonio por defecto de actividad de aldosterona, que ocurre por hipoaldosteronismo o por resistencia a la acción hormonal en los pseudohipoaldosteronismos congénitos o adquiridos. Estos últimos ocurren en lactantes con pielonefritis aguda asociada a anomalías congénitas nefrourológicas y en nefropatías intersticiales con insuficiencia renal crónica, siendo esta la forma más frecuente de ATR tipo 4, en adultos. En las ATR tipo 4 la hiperpotasemia suele ser más llamativa que la acidosis.
ATR distal
Acidosis metabólica hiperclorémica por defecto genético que afecta a la capacidad de acidificación urinaria. Asociación frecuente con sordera nerviosa.
La ATR tipo 1 o distal es la forma de ATR primaria más frecuente en Pediatría. Los pacientes son incapaces de acidificar la orina a un pH ≤ 5,3 en presencia de acidosis metabólica mantenida. La mayor parte de los casos son debidos a defectos de función de los genes ATP6V0A4 o ATP6V1B1, que codifican subunidades de la bomba de protones ATPasa H+ localizada en el borde epitelial de las células del tubo colector o, con menor prevalencia en nuestro medio, del gen SLC4A1 que codifica el intercambiador de aniones que a través de la membrana basal recupera bicarbonato. Hay otros genes causales, pero son mucho más raros, descritos en muy pocos casos. La tabla II muestra características clínicas útiles para el diagnóstico diferencial entre estas tres formas de ATR distal, debiendo confirmarse con estudio genético(8).
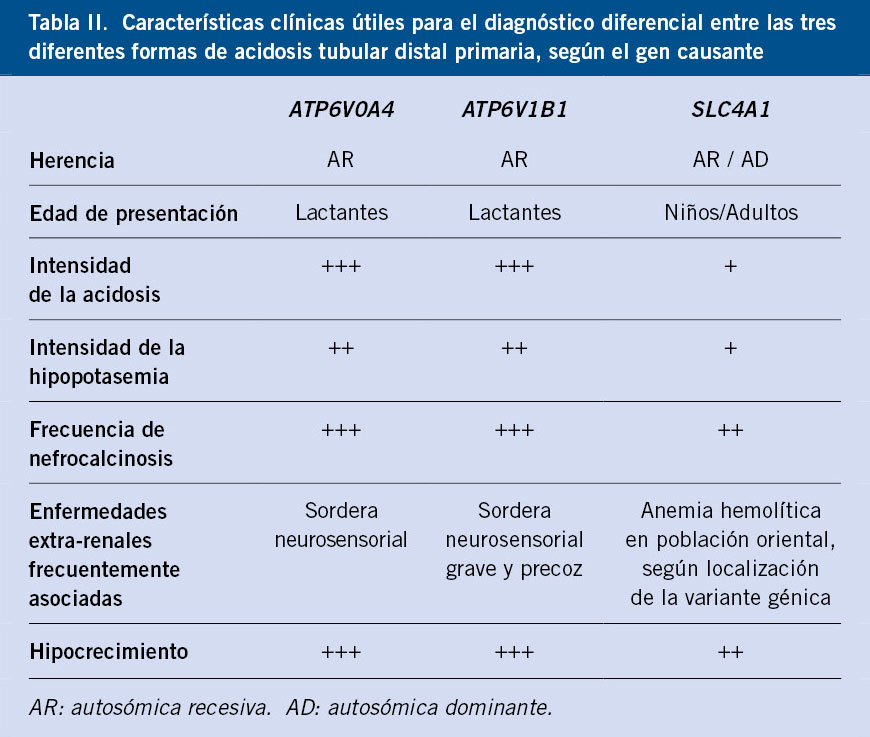
La nefrocalcinosis de la ATR distal es muy precoz y se produce por la asociación de hipercalciuria e hipocitraturia. Estos factores también entrañan riesgo de urolitiasis, aunque esta es más tardía y evitable en gran medida con un adecuado tratamiento que el lector puede consultar en una guía clínica reciente(9). La base de tratamiento es la administración bicarbonato sódico y/o citrato potásico para normalizar el pH sanguíneo y la bicarbonatemia. Las dosis no son altas, entre 2-3 mEq/kg/día, ya que no hay pérdida proximal de bicarbonato, salvo en lactantes al diagnóstico, siendo entonces necesario administrar transitoriamente dosis mayores. Un correcto tratamiento mejora el crecimiento y previene la progresión de la nefrocalcinosis. Sin embargo, estudios recientes indican que un 30 % de pacientes o más desarrollan a largo plazo insuficiencia renal crónica leve-moderada(10).
Equilibrio hidroelectrolítico
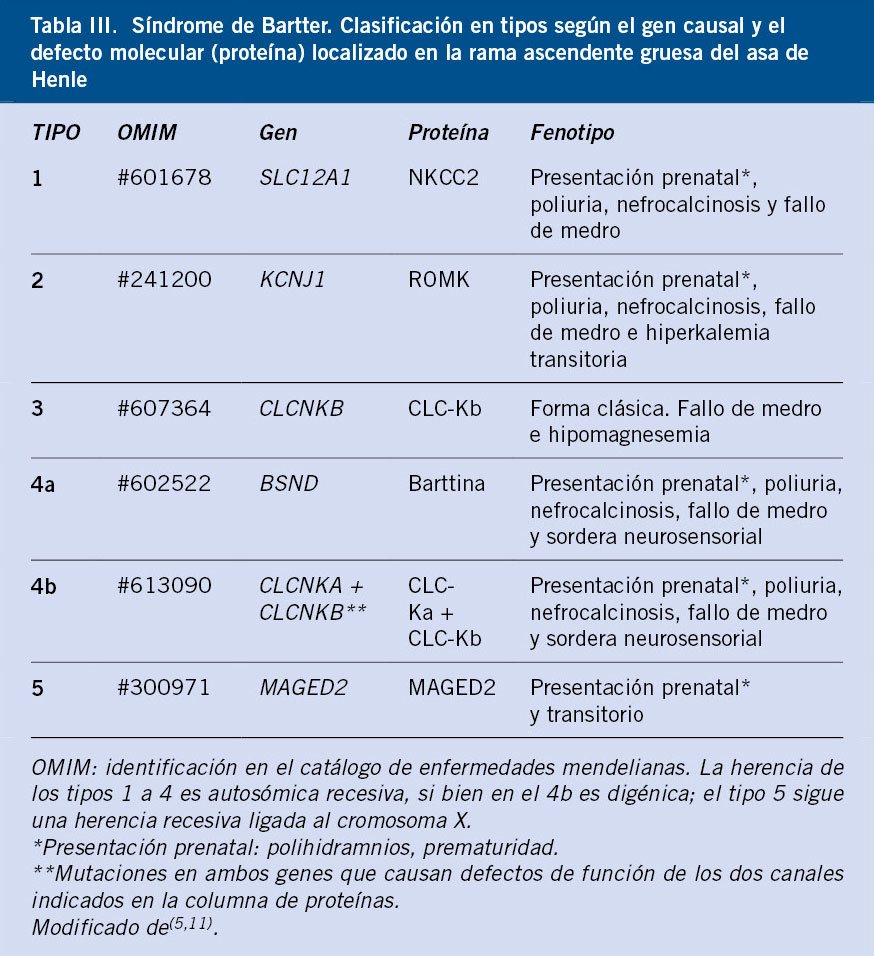
La diabetes insípida nefrogénica (DIN) es la tubulopatía más representativa relacionada con el metabolismo del agua, mientras que en relación con el metabolismo sodio-potasio, las tubulopatías pierde sal más ilustrativas son el síndrome de Bartter, que engloba diversas entidades cuyas características principales se resumen en la tabla III(5,11) y el síndrome de Gitelman.
Diabetes insípida nefrogénica
Poliuria masiva en varones por resistencia a la acción de la hormona antidiurética con polidipsia secundaria. Marcada hipostenuria con incapacidad de concentrar la orina por encima de la osmolalidad plasmática.
La DIN está causada por la incapacidad de los túbulos colectores para reabsorber agua por resistencia a la acción de la hormona antidiurética (ADH) o vasopresina (VP). La forma genética más prevalente de DNI, que supone un 90 % de casos, se presenta en varones con un defecto en el receptor V2 de la VP (OMIM 304800) de transmisión recesiva ligada al cromosoma X. Un 10 % de pacientes de ambos sexos tienen DIN de herencia autosómica dominante o recesiva causada por defectos de función en el canal de agua acuaporina-2.
La DIN de origen genético cursa con marcada poliuria, hipostenuria, con osmolalidades urinarias muy inferiores a la plasmática, y polidipsia secundaria con gran avidez por el agua. Si se limita el acceso a los líquidos, los niños presentan episodios graves de deshidratación hipernatrémica. La sed provoca irritabilidad y la alta ingesta hídrica puede dificultar el aporte nutricional y ocasionar déficit de medro, así como hidronefrosis a medio-largo plazo por el alto volumen de orina.
Al igual que en las DIN adquiridas, por nefropatía tubulointersticial crónica o tratamiento por litio en adultos, hay formas parciales de DIN congénita en las que la poliuria y el defecto de concentración urinaria son menos intensos y tienen mucha menor repercusión clínica(12).
Se describe clásicamente el test de deprivación hídrica y administración de desmopresina para el diagnóstico diferencial de la poliuria-polidipsia en Pediatría. En la DIN, la osmolalidad urinaria se mantiene por debajo de la plasmática tras la restricción hídrica y aumenta menos de un 50 % con desmopresina. En los pacientes con DIN primaria este test es muy mal tolerado y entraña riesgo de deshidratación e hipovolemia graves. Debe de realizarse bajo monitorización hospitalaria estrecha y solo si es estrictamente necesario y no puede llegarse a un diagnóstico cierto con la historia clínica, el estudio genético y, recientemente, la determinación de copeptina circulante, un péptido estable, fácil de medir y que es un biomarcador fiable de los niveles de ADH(13).
El tratamiento de la DIN se basa en las medidas ya señaladas: líquidos orales a demanda, dietas bajas en sodio, reducción del filtrado glomerular mediante antiinflamatorios no esteroideos y administración de tiazidas con diuréticos ahorradores de potasio. En la actualidad se están ensayando nuevos fármacos en la DIN primaria dirigidos a contrarrestar específicamente el defecto molecular subyacente(12).
Síndrome de Gitelman
Tubulopatía primaria más prevalente, especialmente en etnia gitana. Nefropatía pierde sal con alcalosis metabólica hipopotasémica, hipomagnesemia e hipocalciuria.
El síndrome de Gitelman (OMIM 263800) es de herencia AR, causado por un defecto en el gen SLC12A3 que codifica el cotransportador de cloruro sódico NCC situado en el túbulo distal. Es posiblemente la tubulopatía primaria más prevalente, con alta incidencia en la etnia gitana(14).
Se presenta característicamente en la infancia tardía o edad adulta y cursa bioquímicamente por alcalosis metabólica hipopotasémica con hipomagnesemia e hipocalciuria. Además de por estas alteraciones analíticas frecuentemente apreciadas por primera vez en el curso de un cuadro de vómitos e intolerancia oral, se puede detectar en individuos asintomáticos en el curso de un estudio familiar, por la avidez por alimentos salados o por clínica neuromuscular consistente en calambres, debilidad, parestesias o tetania. Es una tubulopatía pierde sal y fenotípicamente puede ser indistinguible del síndrome de Bartter tipo 3 (Tabla III), aunque la baja eliminación de calcio con calcio/creatinina inferior a 0,05 mg/mg es un hallazgo muy típico de síndrome de Gitelman, útil en el diagnóstico diferencial clínico(14).
El tratamiento se basa en el aporte de dieta rica en sal, evitar periodos prolongados de ayuno y en suplementos, si son necesarios, de cloruro potásico y de magnesio. Existe una guía clínica de consenso recientemente publicada(15).
Metabolismo mineral: calcio – fósforo – magnesio
En este grupo son destacables el raquitismo hipofosfatémico ligado al cromosoma X y el síndrome de hipomagnesemia hipercalciuria, tubulopatías muy significativas, que pueden ser graves y requieren ser diagnosticadas precozmente por el pediatra.
Raquitismo hipofosfatémico ligado a X
Raquitismo hereditario más común por hipofosfatemia primaria causada por exceso de FGF23 que interfiere con la reabsorción de fosfato y la síntesis de 1,25 dihidroxivitamina D en el túbulo renal proximal.
Esta enfermedad, denominada también hipofosfatemia ligada a X (XLH), forma parte de los raquitismos que no se producen por una carencia de vitamina D. Es el raquitismo heredado y la hipofosfatemia de base genética causada por exceso de FGF23 más frecuente(16). El exceso de producción por los osteocitos de FGF23 se produce por un defecto de función del gen PHEX por un mecanismo no completamente aclarado. El aumento de FGF23 inhibe la reabsorción de fosfato, lo que ocasiona hiperfosfaturia, y la síntesis de 1,25(OH)2D, factores ambos que causan defecto de mineralización del hueso dando lugar a raquitismo en edad pediátrica y a osteomalacia en los adultos.
La enfermedad sigue un patrón de herencia dominante ligada a X (OMIM #307800), tiene una amplia variabilidad fenotípica y produce en niños raquitismo, deformidades en los huesos que soportan peso e hipocrecimiento disarmónico con mayor afectación del segmento inferior del cuerpo. La talla baja no se explica únicamente por la hipofosfatemia ni porque las extremidades inferiores se incurven, sino que el aumento de FGF23 puede jugar también un papel directo cardinal sobre la placa de crecimiento. Asimismo, la XLH causa defectos del esmalte y abscesos dentarios, dolores óseos, impotencia funcional de la unidad osteoarticular, síndrome de Arnold-Chiari, y a largo plazo en adultos hipoacusia por afectación de los huesos del oído interno y lesiones de entesopatía(16).
El tratamiento ha consistido tradicionalmente en la administración de suplementos de fosfato y metabolismos de la vitamina D. Sin embargo, la mala tolerancia, el riesgo de inducir toxicidad y la falta de eficiencia para corregir o prevenir manifestaciones importantes de la enfermedad justifican la administración de burosumab, un anticuerpo monoclonal que antagonista los efectos de FGF23 y que ha demostrado en los últimos años ser un avance terapéutico prometedor. El lector interesado puede consultar diversas guías recientes para el diagnóstico, control y tratamiento de los pacientes con XLH(17,18).
Síndrome de hipomagnesemia familiar con hipercalciuria
Disminución de la absorción paracelular de calcio y magnesio en el asa ascendente de Henle por defecto genético en las claudinas 16 o 19. Asociación de alteraciones oculares y riesgo de evolución a fallo renal avanzado.
Tubulopatía de herencia AR causada por defectos en la función de claudina 16 (OMIM #248250) o de claudina 19 (OMIM #307800), proteínas que regulan el transporte de ion calcio e ion magnesio a través de las uniones paracelulares del asa ascendente de Henle.
Los enfermos tienen hipomagnesemia por fuga renal de magnesio, hipercalciuria, nefrocalcinosis, polidipsia y poliuria. Pueden ser hipocrecidos, tener urolitiasis con infecciones urinarias de repetición y evolucionar a fallo renal crónico. Los pacientes con defecto de la claudina 19 tienen característicamente afectación ocular consistente en miopía maligna, coloboma macular y nistagmus(19). La mayoría de los enfermos españoles tienen la misma mutación en el gen CLDN19. La enfermedad no tiene tratamiento específico dirigido a su base patogénica. La hipercalciuria es intensa y muy resistente a las medidas terapéuticas habituales.
Metabolismo del ácido úrico
En este grupo deben de mencionarse las hipouricemias renales, tipo 1 y 2 (OMIM #220150 y #612076, respectivamente). Está afectada la reabsorción de ácido úrico en la membrana luminal de las células tubulares proximales por función defectuosa del intercambiador renal de urato-anión URAT1 (tipo 1) o de un transportador de urato de alta capacidad llamado GLUT9 (tipo 2). Son de herencia autosómica, generalmente AR, cursan con hipouricemia e hiperuricosuria y pueden ser asintomáticas o cursar con urolitiasis o con fallo renal agudo inducido por el ejercicio. No existe tratamiento específico, debiendo procurarse la hidratación y la administración de citrato potásico para mantener el pH urinario entre 6-7(20). Hay una guía clínica recientemente publicada para consulta(21).
Tubulopatías que afectan todos los sistemas metabólicos anteriores
Síndrome de Fanconi
Afectación global de los procesos de reabsorción en el túbulo proximal. La forma genética más frecuente es la causada por la nefropatía cistinótica.
El síndrome de Fanconi o de Toni-Debré-Fanconi corresponde a una disfunción generalizada del proceso de reabsorción en el túbulo proximal, lo que ocasiona las alteraciones mostradas esquemáticamente en la figura 1.

Figura 1. Síndrome de Fanconi: alteraciones bioquímicas características.
No se trata de un defecto molecular de los múltiples transportadores y canales que intervienen en la reabsorción, sino de un “colapso” global y no selectivo de la función reabsortiva por mecanismos no bien conocidos, entre los que puede existir un daño extenso de la integridad de las células tubulares proximales o una imposibilidad para utilizar energía por las mismas por mal funcionamiento de la ATPasa Na+/K+ o por patología mitocondrial, bien directamente o por lesión de otra organela intracelular(22).
El síndrome de Fanconi puede ser adquirido por tóxicos, medicamentos o enfermedades inmunológicas o puede ser congénito, en el contexto de errores innatos del metabolismo, enfermedades mitocondriales, defectos genéticos del túbulo proximal, enfermedad de Wilson o idiopático. La causa congénita del síndrome de Fanconi más frecuente es la cistinosis (OMIM #219800), enfermedad de depósito lisosomal de cistina causada por defecto del gen CTNS(23).
El síndrome de Fanconi se puede instaurar progresivamente y, cuando es completo, produce manifestaciones clínicas graves compatibles con el perfil clínico descrito de las tubulopatías, además de las dependientes de la enfermedad primaria y requiere las medidas terapéuticas generales ya comentadas. En el caso de la cistinosis existe un tratamiento específico con cisteamina, un agente depletante de cistina que, si bien mejora enormemente el pronóstico de la enfermedad, no es eficaz para controlar la tubulopatía proximal.
Enfermedad de Dent
Tubulopatía proximal en varones con proteinuria de bajo peso molecular, hipercalciuria y riesgo elevado de fallo renal crónico.
Tubulopatía proximal compleja con herencia ligada al cromosoma X causada por alteración en el proceso de endocitosis, clave para la reabsorción de proteínas por defecto de función del gen CLCN5, Dent tipo 1 (OMIM #300009), o del gen OCRL, Dent tipo 2 (OMIM #300555); si bien, en un 25-35 % de los casos no se evidencian mutaciones en ninguno de estos dos genes(24). Los pacientes son varones con pérdida de proteínas de bajo peso molecular (99 % de los casos), hipercalciuria (75-90 %), urolitiasis (30-50 %), nefrocalcinosis (40-75 %) y fallo renal crónico (del 30 al 80 % en mayores de 30 años). En la enfermedad de Dent 2 puede haber casos con afectación oftalmológica y neurológica, con un fenotipo menos grave que el del síndrome de Lowe.
Síndrome de Lowe
También llamado síndrome óculo-cerebro-renal (OMIM #309000). Se afecta el gen OCRL en exones diferentes a la enfermedad de Dent tipo 2. También tiene herencia ligada al cromosoma X y clínicamente cursa con: patología ocular (cataratas, glaucoma, microftalmos, queloides corneales), grave afectación neurológica (hipotonía, arreflexia, retraso cognitivo, alteraciones de conducta, convulsiones) y tubulopatía proximal con hiperaminoaciduria que no progresa siempre a un síndrome de Fanconi completo. Los pacientes pueden progresar a enfermedad renal terminal a los 30-40 años de edad(24).
Función del pediatra de Atención Primaria
Las tubulopatías primarias son enfermedades complejas y, si bien individualmente son de muy baja prevalencia, representan en su conjunto una parte muy significativa y típica de las enfermedades del niño. Es preciso conocerlas para poder diagnosticarlas pronto y evitar o minimizar las complicaciones. Como se muestra gráficamente en el algoritmo que acompaña a este artículo, el pediatra de Atención Primaria debe de:
• Sospechar la presencia de una tubulopatía en un niño, frecuentemente lactante y de corta edad, que presenta déficit de medro o hipocrecimiento con episodios de desequilibrio hidroelectrolítico y ácido-base recurrentes o no aparentemente justificados, así como signos radiológicos o bioquímicos de raquitismo no nutricional.
• Realizar la historia familiar y un árbol genealógico que permita averiguar la existencia de enfermedades de transmisión autosómica, generalmente recesiva, o ligada al cromosoma X, ya que la variabilidad fenotípica de las tubulopatías es muy amplia, incluso dentro de una misma familia, y el estudio familiar conduce en un número significativo de casos al diagnóstico en individuos asintomáticos o con mínima expresividad clínica.
• Interpretar adecuadamente los estudios básicos bioquímicos de la función renal para poder detectar anomalías que justifiquen un estudio diagnóstico de tubulopatía más especializado.
• Contribuir al seguimiento ambulatorio de estos pacientes, detectando complicaciones, minimizando el número de ingresos hospitalarios y orientando a los padres y al niño en aspectos pronósticos y terapéuticos.
Conflicto de intereses
No hay conflicto de interés en la elaboración del manuscrito.
Bibliografía
Los asteriscos reflejan el interés del artículo a juicio del autor. Se han marcado con *** aquéllas que corresponden a capítulos o monografías de alta calidad que revisan la valoración clínica de la función tubular y las tubulopatías. Asimismo, se han resaltado con ** guías clínicas de interés y con * artículos o capítulos de revisión o actualización.
1.*** Hernández Marco R, García Nieto VM, Fons Moreno J. Evaluación de la función renal y tubulopatías. Villanueva de la Cañada: Comunicación y Ediciones Sanitarias S.L. 2021.
2.*** Santos F, García Nieto V. Función renal basal. En: García-Nieto V, Santos Rodríguez F, Rodríguez-Iturbe B, ed. Nefrología Pediátrica, 2nd ed. Madrid: Grupo Aula Médica; 2006. p. 39-49.
3.*** Marín Serra J, Ferrando Monleón S. Valoración de la función renal. En: Antón M, Rodríguez LM, ed. Nefrología Pediátrica. Manual práctico. Madrid: Editorial Médica Panamericana, S.A.; 2011. p. 57-63.
4.* Downie ML, López García SC, Kleta R, Bockenhauer D. Inherited tubulopathies of the kidney: insights from genetics. Clin J Am Soc Nephrol. 2021; 16: 620-30.
5. Kleta R, Bockenhauer D. Salt-losing tubulopathies in children: what’s new, what’s controversial? J Am Soc Nephrol. 2018; 29: 727-39.
6. Barbosa M, Lopes A, Mota C, Martins E, Oliveira J, Alves S, et al. Clinical, biochemical and molecular characterization of cystinuria in a cohort of 12 patients. Clin Genet. 2012; 81: 47-55.
7.* Gil-Peña H, Mejía N, Santos F. Renal tubular acidosis. J Pediatr. 2014; 164: 691-8.
8.* Alexander RT, Law L, Gil-Peña H, Greenbaum LA, Santos F. Hereditary distal renal tubular acidosis. En: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, ed. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 2019. p. 1993-2022.
9.** Trepiccione F, Walsh SB, Ariceta G, Boyer O, Emma F, Camilla R, et al. Distal renal tubular acidosis: ERKNet/ESPN clinical practice points. Nephrol Dial Transplant. 2021; 36: 1585-96.
10. López-García SC, Emma F, Walsh SB, Fila M, Hooman N, Zaniew M, et al. Treatment and long-term outcome in primary distal renal tubular acidosis. Nephrol Dial Transplant. 2019; 34: 981-91.
11.* Walsh PR, Tse Y, Ashton E, Iancu D, Jenkins L, Bienias M, et al. Clinical and diagnostic features of Bartter and Gitelman syndromes. Clin Kidney J. 2018; 11: 302-9.
12.* Moeller HB, Rittig S, Fenton RA. Nephrogenic diabetes insipidus: essential insights into the molecular background and potential therapies for treatment. Endocr Rev. 2013; 34: 278-301.
13. Refardt J, Winzeler B, Christ-Crain M. Copeptin and its role in the diagnosis of diabetes insipidus and the syndrome of inappropriate antidiuresis. Clin Endocrinol (Oxf). 2019; 91: 22-32.
14.* Santos Rodríguez F, Coto García E. Síndrome de Gitelman. En: Exeni R, García-Nieto V, Medeiros M, Santos F, ed. Nefrología Pediátrica. Oviedo: Ediciones de la Universidad de Oviedo; 2021. p. 237-40.
15.** Blanchard A, Bockenhauer D, Bolignano D, Calò LA, Cosyns E, Devuyst O, et al. Gitelman syndrome: consensus and guidance from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2017; 91: 24-33.
16.* Bitzan M, Goodyer PR. Hypophosphatemic rickets. Pediatr Clin North Am. 2019; 66: 179-207.
17.** Haffner D, Emma F, Eastwood DM, Duplan MB, Bacchetta J, Schnabel D, et al. Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nat Rev Nephrol. 2019; 15: 435-55.
18.** Padidela R, Cheung MS, Saraff V, Dharmaraj P. Clinical guidelines for burosumab in the treatment of XLH in children and adolescents: British paediatric and adolescent bone group recommendations. Endocr Connect. 2020; 9: 1051-6.
19.* Claverie-Martín F, Perdomo-Ramirez A, García-Nieto V. Hereditary kidney diseases associated with hypomagnesemia. Kidney Res Clin Pract. 2021; 40: 512-26.
20. Peris Vidal A, Marin Serra J, Lucas Sáez E, Ferrando Monleón S, Claverie-Martín F, Perdomo Ramírez A, et al. Hipouricemia renal hereditaria tipo 1 y 2 en tres niños españoles. Revisión de casos pediátricos publicados. Nefrología. 2019; 39: 355-61.
21.** Nakayama A, Matsuo H, Ohtahara A, Ogino K, Hakoda M, Hamada T, et al. Clinical practice guideline for renal hypouricemia (1st edition). Hum Cell. 2019; 32: 83-7.
22.* Forero Delgadillo JM, Santos Rodríguez F. Tubulopatías proximales complejas. Síndrome de Toni-Debré-Fanconi. Nefropatía por cistinosis. En: Exeni R, García-Nieto V, Medeiros M, Santos F, ed. Nefrología Pediátrica. Oviedo: Ediciones de la Universidad de Oviedo; 2021. p. 169-76.
23.* Elmonem MA, Veys KR, Soliman NA, van Dyck M, van den Heuvel LP, Levtchenko E. Cystinosis: a review. Orphanet J Rare Dis. 2016; 11: 47.
24.* Ehlayel AM, Copelovitch L. Update on Dent disease. Pediatr Clin North Am. 2019; 66: 169-78.
Bibliografía recomendada
– Exeni R, García-Nieto V, Medeiros M, Santos F, ed. Nefrología Pediátrica. Oviedo: Ediciones de la Universidad de Oviedo; 2021.
Es un tratado reciente sobre la fisiología y patología renal en niños y adolescentes. Escrito en español por diversos autores españoles e hispanoamericanos, se edita conjuntamente en España y Méjico y constituye el libro básico de referencia sobre Nefrología Pediátrica para la comunidad médica hispanoparlante en todo el mundo.
– RenalTube: www.renaltube.com
Es un recurso disponible por Internet en el que se puede encontrar información sobre diversas tubulopatías en las que trabajan grupos españoles y se pueden incluir pacientes para su diagnóstico genético.
– Luis Yanes MI, García García PM, García Nieto V. Tubulopatías. 2020.
Artículo de revisión sobre tubulopatías que puede verse y descargarse de la web de la Sociedad española de Nefrología: https://www.nefrologiaaldia.org/253.
| Caso clínico |
|
Niño de 22 meses que ingresa en Urgencias por cuadro febril de infección de vías respiratorias altas. Una tira reactiva en orina evidencia proteinuria y glucosuria, por lo que ingresa para estudio. No llama la atención a los padres que beba grandes cantidades de agua y la diuresis les parece normal. Percentiles 3-10 de peso y talla. Antecedentes familiares: hijo único, padres sanos y no consanguíneos. Antecedentes personales: embarazo a término, peso: 3.850 g; talla: 52 cm; sin enfermedades significativas; correctamente vacunado; convulsión febril simple a los 20 meses de vida. Datos complementarios Sangre/suero: pH 7,41; bicarbonato: 21,5 mEq/l; hemoglobina: 11,9 g/dl; creatinina: 0,50 mg/dl; urea: 24 mg/dl; glucosa: 81 mg/dl; Na: 135 mEq/l; K: 3,92 mEq/l; Cl: 110 mEq/l; calcio: 9,5 mg/dl; fósforo: 2,6 mg/dl; magnesio: 2,23 mg/dl; FA (fosfatasas alcalinas): 1.172 UI/l; urato: 1,1 mg 7 dl. Orina: cromatografía de aminoácidos: aumento de todas las fracciones. Ca/Cr: 1,2 mg/mg (elevado); FG estimado: 92 ml/min/1,73m2 (normal); V %: 3,3 ml/dl FG; EFNa: 2,3 %; EFK: 30,7 %; EFCl: 3,31 %; RTP: 52 %. Rx de huesos largos: ensanchamiento metafisiario con líneas de esclerosis.
|


 Hypercalciuria and hypocitraturia. The concept of prelithiasis in Pediatrics
Hypercalciuria and hypocitraturia. The concept of prelithiasis in Pediatrics