 |
| Temas de FC |
D. González-Lamuño, A. Buendía de Guezala
Unidad de Nefrología-Metabolismo Infantil. Servicio de Pediatría. Hospital Universitario Marqués de Valdecilla. Santander
| Resumen
El Síndrome Nefrítico (SN) se caracteriza por la aparición repentina de edemas e hipertensión arterial (HTA) por efecto de la expansión del sector extracelular hacia los espacios vasculares e intersticiales. La HTA puede complicarse y necesitar tratamiento con hipotensores de acción rápida (diuréticos y antagonistas de calcio). De forma paralela, existe oliguria con orina hematúrica y presencia de proteinuria, con natriuresis baja. A menudo, se acompaña de insuficiencia renal moderada, que en algunos casos puede ser grave y necesitar depuración extrarrenal. Estos síntomas se presentan como consecuencia de una inflamación de los glomérulos (glomerulonefritis aguda, GNA). En la edad pediátrica, las GNA habitualmente se presentan en un contexto clínico postinfeccioso (GNAPI) o como nefritis asociada a una vasculitis por inmunoglobulina A (VIgA). El cuadro más habitual se debe a una GNAPI cuya patogenia es inmune, afecta principalmente a niños entre 2 y 12 años, y tiene una evolución favorable. Además de la hematuria, edemas, oliguria e HTA, existe hipocomplementemia y un antecedente infeccioso reciente o actual. El sedimento urinario suele estar alterado, con proteinuria de intensidad variable, raramente en rango nefrótico. Si se trata de una GNA rápidamente progresiva o con semilunas difusas, el pronóstico no es tan favorable, con HTA poco significativa, pero con insuficiencia renal grave. Una anamnesis minuciosa ofrece, por lo general, elementos diagnósticos sugerentes. |
| Abstract
Nephritic Syndrome (NS) is characterized by the sudden appearance of edema and arterial hypertension (HT) due to the expansion of the extracellular sector towards the vascular and interstitial spaces. If hypertension complicates treatment with rapid-acting hypotensives (diuretics and calcium antagonists) will be required. In parallel, there is presence of oliguria with hematuric urine and proteinuria, with low natriuresis. It is often accompanied by moderate renal failure, which in some cases may be severe and require extrarenal clearance. These symptoms occur as a consequence of inflammation of the glomeruli (acute glomerulonephritis, AGN). In the pediatric age, AGN usually manifests in a post-infectious clinical context (PIAGN), or as nephritis associated with immunoglobulin A vasculitis (IgAV). The most common presentation is due to a PIAGN whose pathogenesis is immune, mainly affects children between 2 and 12 years of age, and has a favorable course. In addition to hematuria, edema, oliguria, and hypertension, there is hypocomplementemia and a recent or current history of infection. The urinary sediment is usually abnormal, with proteinuria of variable degree, rarely in the nephrotic range. If it is a rapidly progressive AGN or with diffuse crescents, the prognosis is not so favorable, with insignificant HT, but with severe renal failure. A careful history usually offers suggestive diagnostic key elements. |
Palabras clave: Síndrome nefrítico; Glomerulonefritis aguda; Insuficiencia renal aguda; Hipocomplementemia; Glomerulonefritis postinfecciosa; Nefropatía IgA.
Key words: Nephritic syndrome; Acute glomerulonephritis; Acute renal failure; Hypocomplementemia; Postinfectious glomerulonephritis; IgA nephropathy.
Pediatr Integral 2022; XXVI (8): 471.e1 – 471.e13
OBJETIVOS
• Comprender que la patogenia de la mayoría de las glomerulonefritis agudas (GNA) en la edad pediátrica es de origen inmune en un contexto infeccioso o asociadas a vasculitis por inmunoglobulina A (IgA).
• Entender en qué consiste el síndrome nefrítico (SN), sus múltiples causas, que la mayoría tienen buen pronóstico, pero que se debe descartar una posible patología sistémica asociada.
• Saber que una adecuada anamnesis junto a la identificación de determinadas variables biológicas (función renal y complemento) permiten orientar las GNA.
• Conocer que la HTA es una de las principales causas de morbimortalidad y que es necesario un adecuado tratamiento.
• Aprender las bases fisiopatológicas de esta enfermedad renal.
Síndrome nefrítico y glomerulonefritis
Introducción
La glomerulonefritis aguda se manifiesta como un síndrome nefrítico de inicio súbito con hematuria, edemas e HTA por expansión del sector extracelular hacia los espacios vasculares e intersticiales, disminución de la filtración glomerular, oliguria y retención de sodio y agua, e insuficiencia renal en grado variable.
El síndrome nefrítico (SN) es una enfermedad renal caracterizada por la presencia de hematuria, proteinuria, oliguria y edemas, con un grado variable de hipertensión arterial (HTA) e insuficiencia renal. Estos síntomas se presentan como consecuencia de una inflamación de los glomérulos del riñón –glomerulonefritis–, manifestada de forma repentina en forma de edemas e HTA, debidos a una expansión del sector extracelular hacia los espacios vasculares e intersticiales. Esta situación produce disminución de la filtración glomerular, con la consiguiente oliguria y retención de sodio y agua, acompañado de edemas, HTA e insuficiencia renal en grado variable. Se caracteriza además por una hematuria glomerular con presencia en el sedimento de hematíes dismórficos y cilindros hemáticos y granulosos, junto a proteinuria en grado variable. La hematuria puede ser esporádica, intermitente o persistente, micro o macroscópica. Por último, señalar que el SN puede ocurrir como un proceso renal aislado o ser parte de una enfermedad sistémica o hereditaria.
Desde el punto de vista etiopatogénico, las GNA pueden ser de tipo primario, o más frecuentemente de tipo secundario, asociadas a una infección o a una enfermedad autoinmunitaria. En la edad pediátrica, las GNA habitualmente se presentan en dos contextos clínicos: 1) como una glomerulonefritis aguda postinfecciosa (GNAPI); y 2) como una nefritis asociada a una vasculitis por inmunoglobulina A (VIgA). No debemos olvidar que existen otras causas, en general de peor pronóstico, en las que será muy importante realizar un diagnóstico adecuado para plantear una posible opción terapéutica específica(1). Además de las GNAPI y las VIgA, en el diagnóstico diferencial de las GNA, deben considerarse: la púrpura de Schönlein-Henoch, el lupus eritematoso sistémico y las vasculitis de pequeños vasos (granulomatosis con poliangeitis y poliangeítis microscópica). La diferenciación de estas entidades se basa en la presencia o no de manifestaciones extrarrenales en el contexto de una enfermedad sistémica, y en los estudios complementarios de laboratorio. El dato diferencial biológico más significativo será el estudio del complemento, que será normal en el caso de la púrpura, y con niveles de C3 y C4 muy disminuidos en el caso del lupus (Tabla I) (Fig. 1). Otras causas posibles de GNA son las GNAPI de etiología diferente a la infección estreptocócica, como son las GNA asociadas a las endocarditis y a los shunts, y la GNA asociada a la hepatitis B.
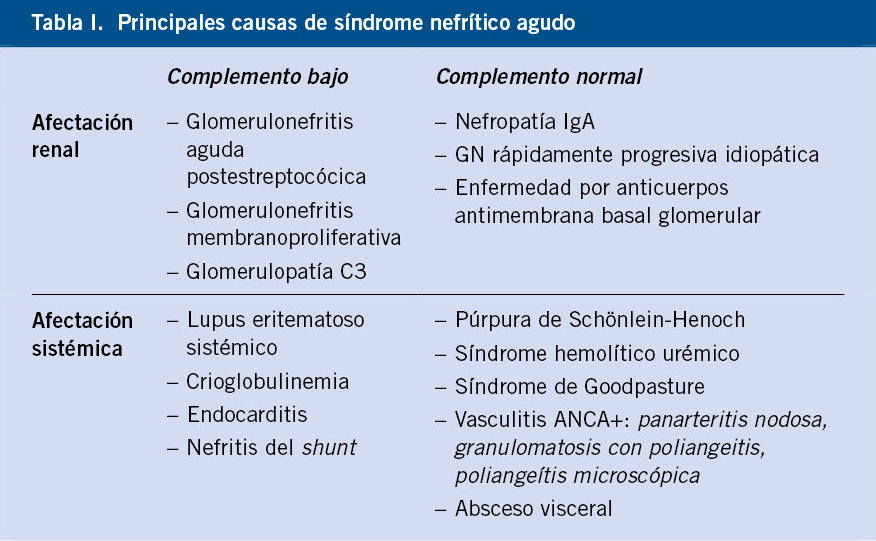
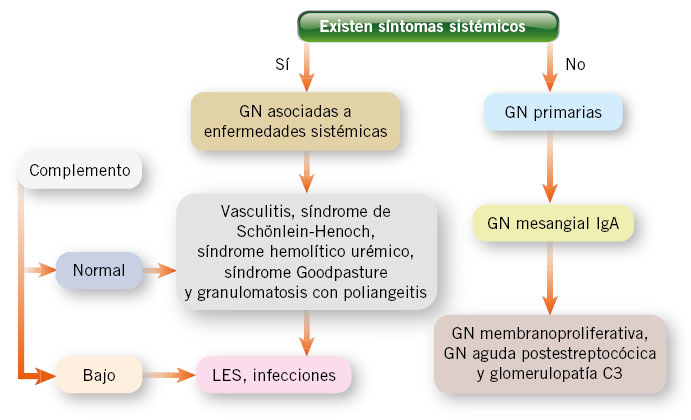
Figura 1. Algoritmo diferencial de las GN en función de la afectación sistémica y alteración del complemento. GN: glomerulonefritis; LES: lupus eritematoso sistémico.
Anamnesis
En un síndrome nefrítico el comienzo es: brusco, con fiebre, cefalea y dolor abdominal; o progresivo, con edema periférico, ganancia de peso y astenia. En ambos casos, está presente una hematuria y la anamnesis debe estar dirigida a diferenciar el origen glomerular o urológico. La hematuria glomerular se describe como color marrón, té o coca-cola, mientras que la urológica se describe como roja encendida y, en ocasiones, con coágulos.
Las manifestaciones de un SN agudo pueden ser: de comienzo brusco, el paciente refiere fiebre, cefalea y dolor abdominal; o progresivo, con edema periférico, ganancia de peso y astenia. El síntoma prínceps suele ser la hematuria, que puede acompañarse o no de oliguria. La orina tiene espuma como resultado de la eliminación de proteínas, que tienen una acción reductora de la tensión superficial de la orina. En la anamnesis al paciente, las preguntas deben estar dirigidas a tratar de diferenciar el origen de la hematuria, bien glomerular, bien urológica o urotelial. La hematuria glomerular se describe como color marrón, té o color coca-cola, mientras que la urológica se describe como roja encendida y, en ocasiones, con coágulos. En la hematuria de origen glomerular, rara vez se recoge la presencia de dolor, y si está presente este es leve en flanco, espalda o abdomen. En la hematuria urológica el dolor es la norma. La pregunta sobre antecedente cercano de fiebre, infección del tracto respiratorio superior o de la piel es obligada. Es además importante recoger antecedentes como: la edad, especialmente relevante si se trata de un niño menor de 4 años o mayor de 15 años; la historia familiar de enfermedades glomerulares o de enfermedad renal de causa no filiada; la posible historia previa de síntomas similares; o identificar signos alarma, como es la evidencia de enfermedad extrarrenal o sistémica, o de una enfermedad renal crónica asociada a HTA.
Examen físico
Los hallazgos físicos más característicos del síndrome nefrítico son el edema periférico y la HTA sistémica, secundarios a la expansión del volumen.
El edema es, en general, moderado, pero el paciente puede llegar a presentar anasarca, insuficiencia cardiaca con edema agudo de pulmón y edema cerebral. Asimismo, la HTA suele ser moderada; pero, en ocasiones, puede presentarse como HTA maligna con daño en órganos diana. Otros hallazgos que pueden orientar a una etiología sistémica o hereditaria del SN, son las alteraciones cutáneas: en forma de rash típico, en ala de mariposa, en el lupus eritematoso sistémico; la presencia de lesiones purpúricas palpables en las vasculitis, entre las que se encuentra la púrpura de Schönlein-Henoch; y la presencia de angioqueratomas en la enfermedad de Fabry. En las articulaciones, la presencia de artritis, hiperelasticidad o rigidez matutina, pueden indicar la presencia de una enfermedad reumática, colagenosis o vasculitis. El resto de la exploración física es inespecífica (Fig. 2).
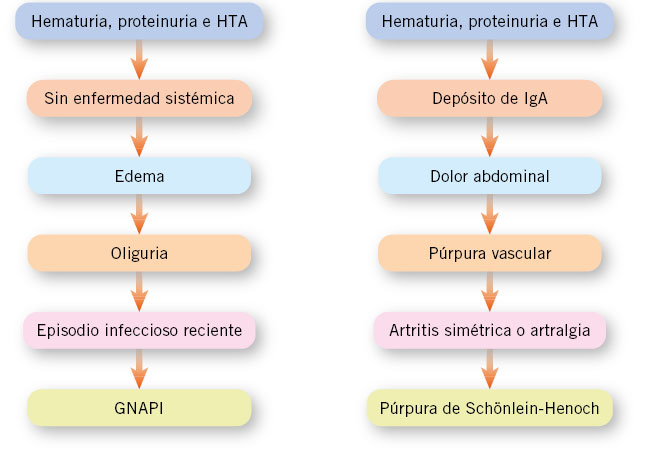
Figura 2. Criterios sugestivos de la glomerulonefritis aguda postinfecciosa (GNAPI) y de la vasculitis por inmunoglobulina A (IgA). HTA: hipertensión arterial.
Fisiopatología del daño glomerular en el síndrome nefrítico (SN)
El síndrome nefrítico es secundario a la inflamación del glomérulo del riñón en el que habitualmente está implicada la respuesta inmunológica. Una vez que se inician los sucesos inflamatorios, aumenta la permeabilidad a las proteínas y la disminución del filtrado glomerular, provocando las alteraciones estructurales del glomérulo con: hipercelularidad, trombosis, necrosis y, en ocasiones, la formación de semilunas.
El SN es secundario a la inflamación del glomérulo del riñón, que puede ser el resultado de alteraciones genéticas primarias, de anomalías en el sistema del complemento o de la coagulación, de disfunciones inmunológicas, o debidos a cambios en la perfusión glomerular. Los trastornos genéticos del glomérulo resultan de mutaciones patogénicas en diferentes genes que codifican para proteínas localizadas, tanto en el glomérulo como en el intersticio y/o en el epitelio tubular. Las alteraciones inmunológicas asociadas a un SN pueden deberse tanto a mecanismos humorales como celulares. Pueden ser debidas a: la presencia de anticuerpos contra los componentes de la estructura del glomérulo, como por ejemplo frente a la membrana basal en la granulomatosis con poliangeitis; la presencia de complejos antígeno-anticuerpo que se escapan al control del sistema retículo endotelial y que, a su vez, se depositan en el glomérulo, como en el caso de la nefropatía IgA; a la interacción antígeno-anticuerpo que se genera y deposita in situ en el propio glomérulo, asociado en ocasiones a liberación de inmunocomplejos circulantes. Otros mecanismos del daño glomerular incluyen disfunciones en el sistema del complemento y de la coagulación, de mecanismos de apoptosis y la síntesis alterada de citocinas, que conllevan la entrada de los leucocitos circulantes. El sistema del complemento se puede activar por la vía clásica, vía lectina o por la vía alternativa. La ruta de activación puede guiar al clínico hacia el diagnóstico subyacente. Una activación por la vía alternativa produce una disminución de los niveles de C3 sérico con niveles de C4 normales, circunstancia característica de la glomerulonefritis aguda postestreptocócica (GNAPE).
Una vez que se han iniciado los sucesos inflamatorios, aparece una cascada de mediadores inflamatorios que son los responsables del incremento de la permeabilidad a las proteínas y de la disminución del filtrado glomerular, provocando las alteraciones estructurales del glomérulo con: hipercelularidad, trombosis, necrosis y, en ocasiones, la formación de semilunas. Se produce un aumento de la reabsorción de sal en la nefrona distal, con la consiguiente retención de líquidos y con un sistema renina-angiotensina-aldosterona que funciona normalmente (Fig. 3)(2).
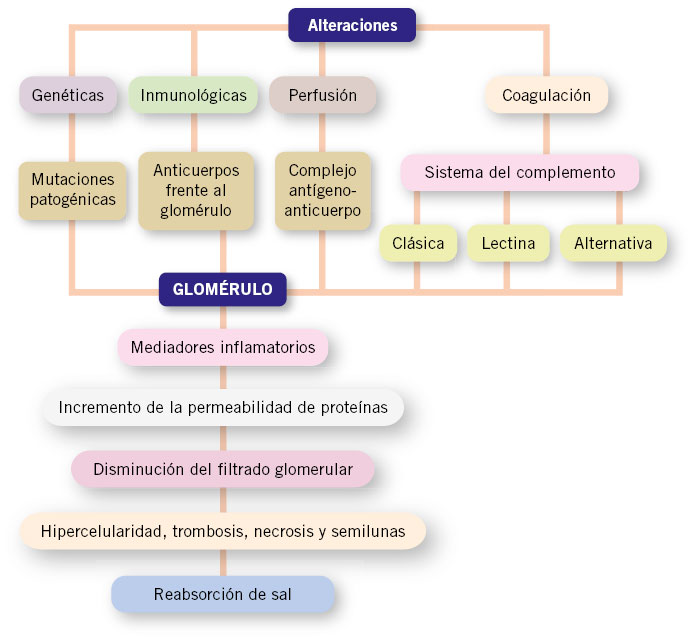
Figura 3. Esquema de los mecanismos fisiopatológicos que actúan en las glomerulonefritis agudas.
Pruebas de laboratorio
Las pruebas de laboratorio básicas, además del estudio del sedimento urinario, incluyen: las pruebas de función renal, el estudio del complemento y de anticuerpos antinucleares y el estudio del sedimento urinario. Una característica identificativa del sangrado glomerular es la presencia de un porcentaje significativo de hematíes dismórficos en el frotis de orina.
Estudios urinarios
La hematuria es fácilmente reconocible, bien macroscópicamente por visión directa o con un resultado positivo para sangre en la tira de orina. La hematuria glomerular se acompaña con frecuencia de proteinuria en grado variable. El rango de la proteinuria puede variar desde bajo grado (<500 mg/día), a una proteinuria en rango nefrótico (>3.000 mg/día). La proteinuria puede cuantificarse mediante la determinación del índice proteinuria/creatinina en orina de una micción aislada, que da una idea bastante aproximada de la magnitud del problema y, si es necesario, mediante la cuantificación en orina de 24 horas. El examen microscópico de la orina en el SN revela un número variable de hematíes libres y, en el caso de orina centrifugada, se observa un botón (pellet) hemático en el fondo del tubo centrifugado. Los cilindros hemáticos son un hallazgo definitivo de afectación glomerular, aunque no se visualizan en todas las ocasiones, ya que requiere visualización en fresco. Pueden aparecer también cilindros granulosos e hialinos, sobre todo si la proteinuria es elevada. Una característica identificativa del sangrado glomerular es la presencia de un porcentaje significativo de hematíes dismórficos en el frotis de orina.
Estudios hemáticos
Los estudios rutinarios de laboratorio incluyen: recuento sanguíneo completo, electrolitos, urea, creatinina y perfil hepático. La velocidad de sedimentación globular (VSG) y la proteína C reactiva están elevadas. La estimación del filtrado glomerular (FG) generalmente se realiza con una recogida de orina de 24 horas para determinar el aclaramiento de creatinina, ya que el FG estimado por la fórmula de Schwartz modificada basa el aclaramiento en una función renal estable, con cifras de creatinina sin variaciones rápidas, y puede arrojar datos erróneos. Ante la sospecha de un SN, es obligado un estudio inmunológico que incluirá complemento y anticuerpos antinucleares (ANA). El C3, componente de la vía clásica y alternativa; y el C4, componente de la vía clásica, permiten aproximar algunos de los mecanismos del trastorno; de esta forma, definiremos el SN con niveles normales de complemento y con niveles alterados (Fig. 1). Niveles bajos de C3 con niveles normales de C4 orientan a una GNAPE o GNAPI, o a una glomerulonefritis membranoproliferativa (GNMP); mientras que niveles bajos de ambos sugieren una GNAPI, un lupus eritematoso sistémico, una GNMP asociada a hepatitis C tipo 1 o una crioglobulinemia mixta. La presencia de ANA positivos nos orienta hacia una enfermedad sistémica. En este caso, otros estudios inmunológicos a incluir serían: anti-ADN, anti-Sm y anti-Ro, para el diagnóstico de enfermedades de colágeno, fundamentalmente el lupus eritematoso sistémico; anticuerpos perinucleares anticitoplasma de neutrófilos y anticuerpos citoplasmáticos anticitoplasma de neutrófilo, para el diagnóstico de vasculitis; y anticuerpos antimembrana basal glomerular, para descartar la granulomatosis con poliangeitis o el síndrome de Googpasture (Tabla I). El SN con C3 bajo puede ser secundario a un número de enfermedades infecciosas, por lo que los estudios deben considerar la política de vacunas de la comunidad, antecedentes personales y edad, serologías de hepatitis virales B y C, sífilis y virus de la inmunodeficiencia humana. Otras enfermedades infecciosas que se deben considerar son: endocarditis; infecciones bacterianas persistentes, como abscesos; o infecciones de shunt vasculares.
Pruebas de imagen
Utilizaremos la ecografía para determinar el tamaño renal y las posibles complicaciones. Aunque un tamaño renal normal no excluye insuficiencia renal crónica, ya que pueden estar aumentados sobre su situación basal debido al SN, los riñones pequeños indican fibrosis irreversible, probablemente atrofia renal.
Biopsia renal
Los pacientes con hematuria de características glomerulares con presión arterial, función renal normal y proteinuria baja no requieren biopsia renal, a menos que sospechemos una enfermedad sistémica con GNA. Las indicaciones absolutas de biopsia renal incluyen: deterioro rápido de la función renal por la sospecha de una glomerulonefritis rápidamente progresiva, insuficiencia renal establecida en la evolución, presencia de proteinuria superior a 1 g/1,73 m2/día, persistencia de proteinuria y alteraciones inmunológicas no compatibles(3).
Desde el punto de vista anatomopatológico, la GNAPI típica es una afección renal secundaria a un episodio infeccioso estreptocócico ocurrido días atrás, con proliferación endocapilar difusa en la biopsia y evolución favorable. La presencia de signos extrarrenales, como exantema cutáneo, fiebre, artralgias o incluso una hemorragia pulmonar, indicará una GNA secundaria asociada a una enfermedad autoinmunitaria. Si se trata de una GNMP, el pronóstico a largo plazo no es tan favorable. El SN agudo puede obedecer también a una glomerulonefritis con semilunas difusas o síndrome de glomerulonefritis rápidamente progresiva (Tabla I). En estos casos, la insuficiencia renal puede ser grave y no remite con rapidez.
Glomerulonefritis aguda postinfecciosa (GNPI)
La glomerulonefritis aguda postinfecciosa es la principal enfermedad glomerular en la edad pediátrica. Consiste claramente en una reacción autoinmunitaria, debida a varias causas, principalmente a agentes microbianos.
Incidencia y distribución estacional
A pesar de ser una condición muy frecuente a nivel global, la incidencia real de la GNAPI, cuyo prototipo en el 90 % de los casos es la GNAPE, es desconocida. La frecuencia de la GNAPE depende mucho del nivel de desarrollo socioeconómico de la región. Se observa una disminución de la incidencia en los países industrializados, debido al mejor acceso a la asistencia sanitaria y a la potabilización del agua de consumo, asociadas a una menor virulencia del estreptococo piógeno. A escala mundial, la mayor parte de los casos ocurren en países de recursos limitados, en los cuales se estima una incidencia entre 10-30 por 100.000 niños, lo que la convierte en la principal enfermedad glomerular en el niño. Existe un cierto predominio en el sexo masculino, y es excepcional en el niño menor de 1 año(4).
La GNAPI puede aparecer de forma esporádica o epidémica, pero con una variación estacional en relación con el foco de infección. En los países occidentales, en los que en general existen variaciones estacionales, es secundaria a las infecciones rinofaríngeas del invierno o la primavera. Por el contrario, en los países tropicales, es secundaria a las infecciones cutáneas y tiende a observarse durante todo el año(5).
Agentes microbianos
El microorganismo más frecuentemente implicado en la GNAPI es el estreptococo beta-hemolítico del grupo A, con un claro predominio del serotipo 12. Además de la proteína de superficie M que permite identificar los serotipos, existen dos grandes grupos de estreptococos del grupo A en función de la presencia o no de una lipoproteinasa que puede opacificar el suero, el denominado factor opacificador del suero; cada uno de ellos contiene un grupo de proteínas M específico. El grupo factor opacificador positivo está formado por cepas nefritogénicas, mientras que el grupo factor opacificador negativo contiene cepas reumatogénicas. Las cepas nefritogénicas se dividen también en cepas responsables de lesiones cutáneas y cepas responsables de infecciones orofaríngeas. Sin embargo, en algunos casos, una cepa asociada a las infecciones cutáneas puede ser responsable de una infección orofaríngea, por transferencia de la cepa de la piel hasta la orofaringe.
Etiología y clasificación
Como se ha mencionado antes, el prototipo de la GNA del niño sigue siendo la GNAPE, aunque existen otros microorganismos implicados en esta enfermedad (Tablas II y III).
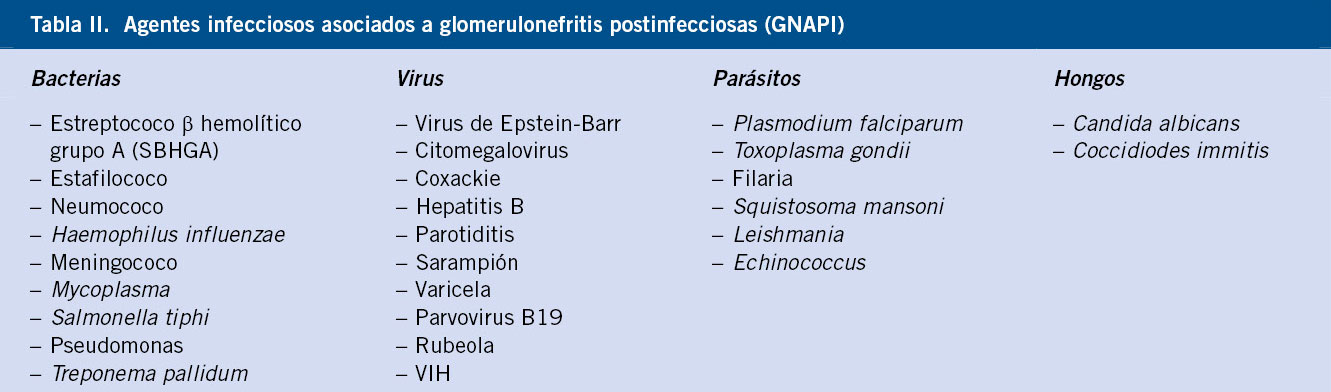

Para sugerir una etiología infecciosa, se deben identificar algunos de los siguientes criterios: un foco infeccioso reciente o actual, ausencia de enfermedad sistémica, IgA escasa o ausente en la biopsia (si se dispone de esta prueba invasiva) y, por último, presencia de lesiones compatibles de origen infeccioso (C3). No hay que olvidar que la GNA puede estar relacionada con una vasculitis (vasculitis por IgA, nefritis secundaria a una endocarditis bacteriana subaguda o una nefritis asociada a un shunt). En ocasiones, una glomerulonefritis crónica se manifiesta en forma de un cuadro nefrítico agudo cuyo diagnóstico diferencial no siempre es fácil de establecer. Este es el caso de las glomerulonefritis primarias asociadas a una nefropatía por IgA o a una GNMP, o en el caso de glomerulonefritis secundaria una nefritis lúpica, por ejemplo.
Cabe mencionar también la glomerulonefritis rápidamente progresiva (GNRP) que es infrecuente en el niño, y los pacientes presentan un cuadro clínico sugestivo de GNA con pérdida progresiva de la función renal en un corto periodo de tiempo (semanas, meses). En la histología de la biopsia renal se observan semilunas difusas.
Patogénesis
La GNA consiste claramente en una reacción autoinmunitaria, debida a varias causas, principalmente a agentes microbianos. Como resultado existe una activación de procesos biológicos que incluyen el reclutamiento de linfocitos T, activación del complemento y liberación de factores de crecimiento y citocinas. Se produce de este modo la reacción inflamatoria y las lesiones glomerulares agudas.
Proceso inmunológico y activación del complemento
Frente a algunos agentes agresores, existe una respuesta inmunitaria humoral con activación del complemento y depósito de inmunoglobulinas. Los antígenos pueden ser componentes de los microorganismos depositados en el glomérulo, como los antígenos catiónicos exógenos de determinadas bacterias (estreptococo, estafilococo), o virus (hepatitis B); o componentes de los propios glomérulos, como el ácido desoxirribonucleico (ADN) del nucleosoma en la nefritis lúpica o los antígenos tumorales. También pueden formarse complejos inmunitarios, circulantes o in situ. En dos tercios de los pacientes con GNA, se han observado complejos inmunitarios circulantes. En el suero de pacientes con GNAPE se demuestra la presencia de valores elevados de inmunoglobulinas G y un título elevado de antiestreptolisina O (ASLO ≥833 unidades Todd).
Posteriormente, se activa la vía alternativa del complemento con la producción de factores quimiotácticos y quimiocinéticos, con depósitos de inmunoglobulina G (IgG), C3, C5 y properdina en los glomérulos. Estos depósitos atraen a los leucocitos hacia el foco de la lesión y facilitan la formación del complejo de ataque de membrana o perforinas. Esta reacción provoca lesiones tóxicas directas en el glomérulo y la liberación de citocinas y prostaglandinas, facilitando la apoptosis celular. La activación de la vía alternativa es todavía más importante por la presencia de un autoanticuerpo estabilizador de la C3-convertasa alternativa, el C3 nefrítico o el C3Nef, pero también por la inhibición del factor H por absorción de la proteína similar al colágeno 1 (Scl-1) del estreptococo. El factor H es uno de los inhibidores fisiológicos de la C3-convertasa alternativa. Asimismo, en algunos casos (GNRP), se puede activar la coagulación formándose trombos y depósito de fibrina que permiten la formación de semilunas.
En la fase inicial de la GNA, la vía clásica del complemento (C1q, C4) y la vía de las lectinas pueden activarse en un menor grado que la vía alternativa. Se ha observado la presencia de anticuerpos dirigidos contra la membrana basal glomerular, la laminina, el colágeno o algunos elementos del mesangio en el suero de pacientes con GNA. Sucede de igual manera con los anticuerpos anti-ADN nativos o los anticuerpos anticitoplasma de los polimorfonucleares (ANCA). Estos últimos aparecen en los casos graves, GNA con insuficiencia renal o GNRP. Se han descrito concentraciones del complemento normales en algunos casos de glomerulonefritis con semilunas. Algunos pacientes pueden presentar lesiones histológicas típicas de la GNAPE sin evidencia de activación del complemento. No se debe olvidar que la fase de hipocomplementemia es muy corta y debe estudiarse de forma precoz. El plazo de normalización del complemento es de 8 semanas, mucho antes de la desaparición de los signos urinarios.
Antígenos estreptocócicos nefritogénicos
• Se trata de un antígeno estreptocócico del grupo A, una enzima glucolítica con actividad gliceraldehído fosfato deshidrogenasa (GAPDH) llamada receptor de plasmina asociado a nefritis (NAPIr). Está demostrado que NAPIr presenta una actividad similar a la plasmina que puede provocar una reacción inflamatoria. Se detecta NAPIr en los depósitos glomerulares del 100 % de los pacientes a los que se realiza una biopsia precoz en una GNAPE.
• Se ha observado también otra proteinasa catiónica exotoxina B (SPeB) en los depósitos subepiteliales de los glomérulos de los pacientes con GNAPE. SPeB es producida por todas las cepas de estreptococo del grupo A y presenta propiedades superantigénicas. Se han observado concentraciones elevadas de anticuerpos anti-SPeB en el suero de pacientes con GNAPE, en comparación con concentraciones más bajas en pacientes con escarlatina, insuficiencia renal aguda o en personas completamente sanas. Asimismo, se ha observado SPeB en muchos de los depósitos glomerulares de los pacientes con GNAPE, frente a un depósito escaso en los pacientes con otras enfermedades glomerulares. Los anticuerpos relacionados con SPeB resultan más útiles que las concentraciones de ASLO y de antiestreptodornasa B (anti-DNasa B) para confirmar una infección causada por una cepa estreptocócica nefritogénica.
• Estudios más antiguos demostraron el papel de la endoestreptocina, una proteína del citoplasma del estreptococo que se ha observado en los glomérulos desde el inicio de la GNAPI. Los anticuerpos dirigidos contra la endoestreptocina están relacionados con la evolución de la enfermedad y podrían utilizarse como marcadores diagnósticos de la GNAPE.
Lesiones glomerulares e inmunidad celular
En las GNA se observa un infiltrado de macrófagos provocado por la activación del complemento, pero también un infiltrado de linfocitos T helper. En consecuencia, existe producción y liberación de sustancias vasoactivas (óxido nítrico, endotelina), citocinas, factores de crecimiento y prostaglandinas. Los factores de crecimiento, como el factor de crecimiento derivado de las plaquetas, el factor de crecimiento transformante beta y el factor de crecimiento endotelial vascular, desempeñan un papel importante en la formación de las lesiones glomerulares, incluida la proliferación de las células glomerulares, el depósito de la matriz extracelular y la esclerosis. En relación con las interleucinas (IL), su papel es evidente. IL-1, IL-8, IL-19 y el factor de necrosis tumoral α (TNF-α) ejercen una acción proinflamatoria. IL-1 provoca la reacción inflamatoria y la proliferación mesangial, mientras que IL-8 es un quimioatrayente para los granulocitos. El número de macrófagos CD16 (que sintetizan el TNF-α) presente en la orina o en los glomérulos es proporcional a la gravedad de la GNA.
Clínica
Tras un intervalo libre variable tras una infección estreptocócica, la GNA se manifiesta como un SN agudo, caracterizado por la aparición súbita de: hematuria, que puede ser macro o microscópica, HTA, retención hidrosódica en forma de edemas y oliguria.
Cabe destacar que la hematuria macroscópica o el edema son los motivos más frecuentes de consulta de los niños con GNAPE y que es excepcional en menores de 2 años(6). La media de edad de aparición oscila entre los 6-8 años, con una proporción por sexos niño/niña de 2. Sin embargo, no existen diferencias en cuanto al sexo en relación con el origen cutáneo u orofaríngeo de la infección estreptocócica. Se han descrito casos subclínicos en la literatura y, hasta un 20 % de los miembros de una familia de un paciente con GNAPI, pueden presentar también la enfermedad. Una hematuria microscópica puede ser el único signo de la enfermedad en un niño cuyo hermano o hermana presenta una forma más florida.
Clásicamente, se describen tres fases en la GNAPE: una fase latente, una fase aguda y una fase de recuperación.
Fase latente
La duración media de la fase latente oscila entre 7-14 días, con un intervalo de 3 a 33 días.
Fase aguda
En la fase aguda, el paciente presenta de forma súbita signos de la afectación glomerular, la retención hidrosódica y signos biológicos. La presencia de sangre en la tira reactiva de orina sugiere la positividad de la hematuria, que se confirmará con el examen microscópico de la orina. Se deben descartar falsos positivos, como por ejemplo, en caso de toma de medicamentos como laxantes o antibióticos (metronidazol) o en caso de coloración anormal de la orina tras ingesta de remolacha. Asimismo, se debe descartar un origen urinario de esta sangre, de la piel, del aparato genital o del recto del paciente. La presencia de hematíes dismórficos en el sedimento de orina, indica el origen glomerular de la hematuria. Con frecuencia, se observan también cilindros hemáticos y leucocitarios. Al inicio de la fase aguda, la leucocituria puede aparecer antes que la hematuria. Existe también proteinuria, generalmente moderada (inferior a 1 g/L), aunque en el 5-25 % de los casos se detecta una proteinuria masiva asociada a un síndrome nefrótico. Algunos autores describen una correlación entre el pronóstico de la función renal y la proteinuria(7).
El segundo signo más frecuente, que aparece en el 64-90 % de los casos, es la HTA que puede estar asociada a graves complicaciones como son: la encefalopatía hipertensiva, una descompensación cardiaca o el edema agudo de pulmón. Existe una buena correlación entre la presión arterial diastólica y el grado de sobrecarga que puede valorarse con la diferencia de peso pre- y post-reanudación de la diuresis. Por lo general, la presión arterial se normaliza en 1-2 semanas y la fracción de excreción de sodio suele ser inferior al 1 %. La encefalopatía hipertensiva descrita en el 30-35 % de los niños con GNAPE, se manifiesta por la presencia de convulsiones, agitación, cefalea, trastornos visuales o alteración del estado neurológico(8).
Generalmente, la actividad de la renina plasmática está disminuida al inicio, con un grado de retención de líquido proporcional a la disminución de la actividad de renina. Por el contrario, la concentración del factor natriurético atrial está elevada en el plasma. Estos datos sugieren una disfunción primaria del túbulo renal.
Algunos pacientes presentan hiperpotasemia relacionada con el túbulo distal, como en un pseudohipoaldosteronismo de tipo 2. La gravedad y la frecuencia de esta hiperpotasemia no se correlacionan con el grado de la insuficiencia renal. Asimismo, a menudo se observa anemia en los pacientes con GNAPE, con frecuencia secundaria a la sobrecarga hídrica. Sin embargo, también se ha descrito una anemia hemolítica autoinmunitaria.
Siempre en la fase aguda, en el 30-45 % de los casos, se observa una insuficiencia renal, de corta duración, inferior generalmente a 3 días. En casos excepcionales (2-3 %), es necesaria la depuración extrarrenal en las formas oligoanúricas.
Para establecer el diagnóstico de la GNAPE, son necesarias la titulación de los anticuerpos antiestreptocócicos: ASLO, anti-DNasa y antihialuronidasa, y la determinación de la fracción C3 del complemento. La ASLO se encuentra más elevada en la GNAPE secundaria a la faringitis que en la forma causada por impétigo, en la que se observan concentraciones elevadas de anticuerpos anti-DNasa B. Es fundamental la repetición de la titulación de los anticuerpos, ya que algunos pacientes con un valor normal de la ASLO pueden presentar en consecuencia un aumento de las titulaciones. La fracción C3 del complemento está descendida y recupera los valores normales a las 6-8 semanas del inicio de la enfermedad. En algunos pacientes, este descenso precede a la aparición de la hematuria(9). En la fase aguda, la ecografía renal puede ser normal o puede mostrar una diferenciación corticomedular, en ocasiones, con un Doppler alterado. La radiografía pulmonar también puede ser normal o mostrar signos de sobrecarga pulmonar en función del cuadro clínico del paciente.
Fase de recuperación
La reanudación de la diuresis con desaparición de la sobrecarga hídrica indica la fase de recuperación. La diuresis puede ser espontánea o estar provocada por los diuréticos, y la presión arterial se normaliza. En esta fase se observa también la desaparición de la proteinuria y de la hematuria macroscópica. En muchos pacientes, la proteinuria desaparece antes que la hematuria microscópica. Se ha sugerido en casos aislados una recidiva de la GNAPE(10). Los autores mencionan la multiplicidad de subtipos de estreptococos que pueden provocar la enfermedad.
Indicaciones y utilidad de la biopsia renal
Indicación
Ante un cuadro clásico de GNAPI, la biopsia renal en general no está indicada. Está recomendada en todo paciente con proteinuria masiva y síndrome nefrótico de más de 7 días de duración o en caso de insuficiencia renal de más de 3 días de evolución en la fase aguda. La biopsia ayuda a descartar una GNA con proliferación extracapilar.
En un segundo tiempo, la biopsia se considerará: en cualquier cuadro de GNA de evolución atípica, en caso de complemento normal en la fase aguda, en caso de proteinuria de más de 8 semanas de evolución o de persistencia de la hematuria microscópica pasados 18 meses. La no normalización de la complementemia a las 8 semanas ha sido considerada durante mucho tiempo como un criterio de biopsia renal con el fin de descartar una GNMP. Para algunos autores, la persistencia de la hipocomplementemia en caso de regresión de los signos clínicos no siempre es considerada como un criterio de exclusión del origen estreptocócico de la GNA. También está indicada la biopsia renal en caso de recidiva de la GNA tras un episodio pasado(11).
Lesiones histológicas
Las lesiones histológicas son fundamentalmente glomerulares; las lesiones tubulointersticiales son transitorias y leves. En la inmunofluorescencia se observan depósitos granulares de C3 y de inmunoglobulinas G en la fase aguda(12). Se pueden encontrar a nivel mesangial puro o en las paredes de los capilares glomerulares de localización extramembranosa, ofreciendo un aspecto de guirnalda, o también se pueden observar de forma difusa a nivel mesangial alrededor de los capilares glomerulares, originando el aspecto clásico de cielo estrellado. Algunos autores han demostrado la existencia del depósito de C3 sin inmunoglobulina G incluso en la fase aguda de la enfermedad en el 30 % de las biopsias. La microscopia óptica muestra una proliferación mesangial y endotelial difusa que afecta a todos los glomérulos(13). Se observa también un infiltrado de la madeja glomerular de células como linfocitos T, neutrófilos y macrófagos. La proliferación endocapilar provoca una reducción de la luz capilar correlacionada con el grado de la insuficiencia renal; una hipocomplementemia importante se asocia también con una proliferación endocapilar intensa. Depósitos extramembranosos eosinófilos de forma cónica rodeados por un halo claro forman las gibosidades. La proliferación extracapilar asociada a medialunas circunferenciales constituye un mal pronóstico. De forma excepcional, se pueden observar medialunas epiteliales en más del 50 % de los glomérulos, provocando un cuadro de glomerulonefritis rápidamente progresiva. Se ha demostrado también una relación entre la ausencia de depósitos paramesangiales y la hipoalbuminemia en los niños con GNAPE. La retención de líquido era mayor en estos pacientes, el complemento normal y la hematuria macroscópica infrecuente. En la microscopia electrónica aparecen depósitos extramembranosos no específicos.
Tratamiento
Tratamiento curativo
El tratamiento del paciente con GNAPI es sintomático, siendo uno de los objetivos principales el manejo de la HTA. La molécula de primera elección son los diuréticos del asa, que se administran asociados a la restricción hidrosódica a dosis de 1-3 mg/kg de peso corporal. En caso necesario, están indicados los antagonistas del calcio si la presión arterial no se controla con la reanudación de la diuresis. Por el contrario, se deben evitar los betabloqueantes en caso de fallo cardíaco.
No se recomiendan tampoco los inhibidores de la enzima convertidora de la angiotensina (IECAS), debido al riesgo de hiperpotasemia e insuficiencia renal. Se pueden proponer inmunosupresores en todo cuadro de GNA grave, glomerulonefritis extracapilar y glomerulonefritis rápidamente progresiva. Algunos autores han llegado a la conclusión de la ausencia de efectos de los inmunosupresores comparados con el tratamiento de soporte en pacientes sometidos a biopsia renal por una evolución clínica atípica. La antibioticoterapia no está indicada salvo presencia de un foco infeccioso activo (dental, pulmonar, cutáneo), y no modifica la historia natural de la GNAPE. La depuración extrarrenal es excepcional.
Tratamiento preventivo
El tratamiento precoz y adaptado de las infecciones estreptocócicas ha permitido observar un claro descenso de la incidencia de la GNAPI en los países industrializados, debido también a la mejora de las condiciones higiénicas y la educación sanitaria de estas comunidades.
Pronóstico
En general, la evolución de la GNA es favorable, sin secuelas renales, con un excelente pronóstico a largo plazo. Sin embargo, las formas con insuficiencia renal grave o una proteinuria importante no tratadas evolucionan, en ocasiones, hacia la fibrosis y la reducción de la cantidad de nefronas. La tasa de mortalidad es baja en los países desarrollados.
En ausencia de un adecuado control clínico, pueden aparecer complicaciones cardiovasculares agudas o una encefalopatía hipertensiva, asociadas a una elevada mortalidad. A los edemas periféricos se pueden sumar: edema agudo de pulmón y edema cerebral, o una HTA grave que puede complicarse con cefaleas, convulsiones con coma y amaurosis. Necesita tratamiento inmediato con hipotensores de acción rápida (inhibidores cálcicos en especial y diuréticos). A menudo se trata de una insuficiencia renal moderada, pero en algunos casos puede ser grave y necesitar depuración extrarrenal.
Los pacientes desarrollan una inmunidad frente a las distintas cepas nefritógenas; sin embargo, se pueden observar una o varias recidivas de GNA en un porcentaje del 1-5 % de los pacientes. La GNAPE con semilunas presenta un mejor pronóstico que la GNA con semilunas por otra causa, aunque no existe unanimidad en relación con este dato(14).
Perspectivas
Se encuentra en fase de evaluación una vacuna multivalente orientada a la región variable de la proteína M bien tolerada en el ser humano, aunque únicamente incluye las cepas reumatogénicas.
Vasculitis por inmunoglobulina A y enfermedad de Berger
La vasculitis por inmunoglobulina A (VIgA), es una forma aguda de la enfermedad de Berger, y en Pediatría representa el grupo de vasculitis primarias más frecuentes junto con la enfermedad de Kawasaki. La VIgA es de aparición estacional y es más frecuente en invierno, con predominio en los varones.
El grupo de edad de 9-15 años es el más afectado. La nefropatía IgA, aunque al igual que la GNAPE puede debutar como un SN, podemos sospecharla en el contexto de un proceso respiratorio, con un intervalo corto, de menos de 5 días de duración, con complemento normal y con un curso clínico en brotes. El paciente puede haber tenido un episodio previo o tenerlo posteriormente, a diferencia de la GNPE, que no suele recidivar. Algunos autores refieren aumento de la IgA sérica, pero en la práctica clínica en los niños es excepcional, encontrándose IgA elevada en menos de un 5 %. La nefropatía IgA cuando comienza como SN agudo y tiene una evolución rápidamente progresiva, se ha intentado tratar con pautas inmunosupresoras, aunque el nivel de evidencia es bajo. Existe un paralelismo de los criterios sugestivos de la glomerulonefritis postinfecciosa y de los de la vasculitis por IgA.
Nefropatía de la púrpura reumática
La púrpura reumática o de Schönlein-Henoch, es una vasculitis sistémica que afecta los vasos de pequeño calibre. Se caracteriza por la combinación de signos y síntomas cutáneos (púrpura), articulares (artralgias) y gastrointestinales (dolores abdominales). Aunque las manifestaciones clínicas son bastante típicas de la enfermedad, el diagnóstico no siempre resulta sencillo por la similitud con el cuadro clínico de otras vasculitis. La lesión glomerular se observa en alrededor del 30 % de los niños y puede ser grave. La enfermedad evoluciona por accesos, caracterizados por una o más manifestaciones clínicas. Las recidivas son más frecuentes durante los tres primeros meses, pero pueden persistir varios años. La lesión renal se puede manifestar o agravar en una de las crisis. Otras manifestaciones clínicas posibles son: pancreáticas, ureterales, testiculares, miocárdicas y neurológicas(15,16).
La púrpura reumática afecta principalmente en edades tempranas (3-15 años) y rara vez en la edad adulta. Es más común en el hombre que en la mujer, con una relación de 1,5-2 a 1. La incidencia anual es de alrededor de 15 casos cada 100.000 niños. A menudo la precede un cuadro respiratorio (infección estreptocócica, adenovirus, parvovirus, Mycoplasma pneumoniae). El mayor número de casos en invierno o primavera confirma el efecto desencadenante de estas infecciones.
Afectación renal
La frecuencia de la lesión renal varía según las series en función del tipo de pruebas empleadas para detectar la nefropatía, de la frecuencia con que estas se han realizado y del modo de selección de los pacientes. En la mayoría de los casos, la lesión renal se produce dentro de los tres primeros meses de evolución, aunque, a veces, puede desarrollarse de manera tardía con ocasión de una recidiva. La gravedad de la nefropatía no es proporcional a la intensidad de las lesiones cutáneas o digestivas. La hematuria microscópica o macroscópica es el signo más habitual y, a menudo, se presenta de forma aislada. Se puede acompañar de proteinuria de abundancia variable con síndrome nefrótico, que solo suele manifestarse por alteración de los parámetros analíticos. La insuficiencia renal es infrecuente al principio y, en general, moderada. En algunos casos se detecta HTA, incluso en niños con manifestaciones urinarias mínimas.
El pronóstico de la nefropatía de la púrpura reumática generalmente es favorable. El porcentaje de niños que progresan hacia la insuficiencia renal terminal no supera el 2-3 % en series no seleccionadas. La magnitud de las manifestaciones clínicas de insuficiencia renal es un buen factor de pronóstico a largo plazo. La curación es lo habitual cuando los signos se reducen a hematuria aislada o asociada a proteinuria inferior a 1 g/24 h. En cambio, puede llegar a la insuficiencia renal si la proteinuria se acompaña de síndrome nefrótico y/o insuficiencia renal, aunque la mitad de los pacientes con esos signos experimentan una evolución favorable a largo plazo. Cada recurrencia de la enfermedad, que en ocasiones se puede manifestar por hematuria macroscópica, podría acompañarse de una agravación de las lesiones histológicas.
Pruebas complementarias
El diagnóstico de púrpura reumática se formula a partir de manifestaciones clínicas cutáneas, articulares y digestivas; las pruebas de laboratorio sirven básicamente para descartar otro diagnóstico y evaluar la intensidad de la lesión renal.
Los valores de las plaquetas son normales. La concentración de IgA plasmáticas aumenta en la mitad de los casos, pero no guarda relación con la gravedad de la enfermedad. Se ha señalado la presencia de factor reumatoideo IgA, complejos inmunitarios IgA y anticuerpos tipo IgA dirigidos contra el citoplasma de los neutrófilos polimorfonucleares (ANCA).
La búsqueda de hematuria y de proteinuria debe realizarse dos veces por semana al comienzo de la enfermedad y después, una vez por semana, durante al menos dos meses, así como mientras persistan las manifestaciones clínicas. Si el resultado de la búsqueda es positivo, debe evaluarse el volumen de la proteinuria, bien en orina de 24 horas, o bien por determinación de la relación proteinuria/creatininuria en una muestra. En caso de incremento de la proteinuria, se indica análisis cuantitativo de proteínas sanguíneas y de albúmina plasmática. La función renal se evalúa a partir de la creatinina plasmática y de la depuración de la creatinina.
El estudio de la biopsia renal en microscopia óptica es muy variable, en lo que se refiere a tipo y gravedad de la lesión glomerular, de un glomérulo a otro y de un paciente a otro. La nefropatía se revela por alteración mesangial con depósitos e hipercelularidad y medialunas epiteliales en grado variable. Según la magnitud de la proliferación celular, pueden identificarse cuatro categorías:
1. Glomerulonefritis mesangiopática con proliferación de células mesangiales.
2. Glomerulonefritis segmentaria y focal de algunos glomérulos, con proliferación en varios ovillos capilares y adherencias a la cápsula de Bowman; los demás glomérulos están indemnes.
3. Glomerulonefritis proliferativa endocapilar con proliferación acentuada y difusa de células mesangiales.
4. Glomerulonefritis endocapilar y extracapilar, en la que a la proliferación mesangial difusa se agregan medialunas epiteliales en un número variable de glomérulos.
El aspecto en inmunofluorescencia es unívoco: en todos los glomérulos existen depósitos granulosos mesangiales que captan de forma predominante el suero anti-IgA, en especial suero anti-IgA1. En las formas histológicas más graves se pueden añadir depósitos endomembranosos. A los depósitos de IgA se asocian muy a menudo depósitos de C3, fibrinógeno e IgG.
Correlaciones anatomoclínicas
En varios estudios se ha demostrado la buena correlación existente entre la magnitud de las manifestaciones iniciales de la lesión renal y la gravedad de la lesión glomerular, evaluada en función del número de glomérulos con medialunas epiteliales. Con respecto al pronóstico a largo plazo, el mejor indicador es el porcentaje de medialunas epiteliales en la biopsia inicial. La mayoría de los pacientes que evolucionaron hacia la insuficiencia renal presentaban en la biopsia inicial, medialunas epiteliales en más del 50 % de los glomérulos. Sin embargo, la evolución en enfermos con lesión histológica grave puede ser favorable, sobre todo cuando el tratamiento se inicia de manera precoz.
Tratamiento
La nefropatía de la púrpura reumática no tiene tratamiento específico. El reposo en cama, prescrito durante mucho tiempo por los pediatras, limita la gravedad de la púrpura, pero no ejerce influencia sobre la lesión renal o digestiva; por tanto, no está realmente justificado.
Se discute el efecto preventivo de la corticoterapia sobre la lesión renal. En algunos estudios aleatorizados realizados en niños que reciben corticoterapia a dosis de 1 mg/kg/día durante 2 semanas, se demuestra la inexistencia de lesión renal en los pacientes tratados y el desarrollo de una nefropatía moderada en el 10-15 % de los niños no tratados. En cambio, esa diferencia no se encontró en estudios retrospectivos(17).
En pacientes con nefropatía se debe considerar la necesidad de indicar tratamiento. Cuando se acompaña de síndrome nefrótico y proliferación extracapilar marcada, se recomiendan 3 perfusiones de metilprednisolona a dosis de 1.000 mg/1,73 m2. Las perfusiones se reemplazan luego con corticoterapia por vía oral. El tratamiento tiene más posibilidades de ser eficaz si se aplica de manera precoz en los primeros meses. Algunos autores han recomendado tratamiento simultáneo con ciclofosfamida por vía oral. El efecto beneficioso de los intercambios plasmáticos en las formas graves no ha sido demostrado(18).
En pacientes que hayan evolucionado hacia la insuficiencia renal, es preferible esperar un año antes de considerar el trasplante renal, siempre que el tiempo de evolución entre el comienzo de la enfermedad y la fase de insuficiencia renal haya sido corto. Después del trasplante y, aunque en el injerto se encuentran muy a menudo depósitos de IgA, es excepcional una recidiva clínica; los resultados del trasplante son idénticos a los alcanzados en todos los niños con trasplante renal por cualquier otra causa.
Glomerulonefritis por depósitos mesangiales de inmunoglobulina A (enfermedad de Berger)
La glomerulonefritis por depósitos mesangiales de IgA o enfermedad de Berger, es la nefropatía glomerular más frecuente en el mundo. En el niño provoca alrededor del 10 % de las nefropatías glomerulares. Afecta más a varones que a mujeres, con una relación de 3/1. Los primeros signos suelen aparecer a los 7-13 años de edad y la enfermedad pocas veces se manifiesta antes de los 4 años(19).
Manifestaciones clínicas
Los signos de presentación de la enfermedad de Berger son variables: desde hematuria aislada hasta insuficiencia renal rápidamente progresiva. La frecuencia relativa de los distintos cuadros clínicos difiere en niños y adultos. En la infancia, la enfermedad se manifiesta por hematuria macroscópica en el 80 % de los casos. Otras veces, la enfermedad se descubre a partir de un análisis de orina rutinario, que revela hematuria microscópica y/o proteinuria. De manera excepcional, asocia un síndrome nefrótico desde el principio. No obstante, algunos pacientes presentan proteinuria abundante con síndrome nefrótico y el análisis de la biopsia renal muestra lesiones glomerulares mínimas o presencia de depósitos mesangiales de IgA. Los pacientes suelen responder a una corticoterapia y la evolución es idéntica a la de los que padecen nefrosis. Es probable que en realidad tengan nefrosis y que a esta se agreguen depósitos de IgA. La concentración de IgA plasmáticas aumenta solo en la mitad de los casos, pero este dato no tiene valor pronóstico(19).
La evolución se caracteriza por la recurrencia de la hematuria macroscópica, que a menudo se produce dentro de las 48 horas siguientes a un cuadro infeccioso, en especial rinofaríngeo. Junto con la hematuria macroscópica puede haber dolor abdominal o lumbar y, en contadas ocasiones, insuficiencia renal aguda transitoria. Esta guardaría relación con una toxicidad tubular de la hemoglobina, ya que las lesiones glomerulares no explican el descenso de la filtración glomerular. La precocidad de la hematuria en relación al cuadro infeccioso y la ausencia de HTA son dos argumentos de peso para descartar una GNAPI. Por añadidura, la fracción C3 del complemento es normal. Los análisis de orina suelen ser normales entre los episodios de hematuria macroscópica. Sin embargo, algunos niños siguen con hematuria microscópica y, a veces, proteinuria. La HTA puede manifestarse, aunque no haya insuficiencia renal. Excepcionalmente, la enfermedad avanza con rapidez hacia la insuficiencia renal terminal en pocos meses.
Anatomía patológica
Como en la púrpura reumática, el daño glomerular se caracteriza por alteración mesangial con depósitos, además de hipercelularidad y medialunas epiteliales en grado variable. El diagnóstico de enfermedad de Berger solo se consigue mediante análisis en inmunofluorescencia. Así se revelan depósitos que captan el suero anti-IgA de manera predominante. A esos depósitos se suman a menudo otros de IgG, IgM, C3 y fibrina con idéntica localización. La mayoría de las veces se trata de depósitos puramente mesangiales, pero en las formas graves, los depósitos de IgA también se encuentran en las paredes de los capilares glomerulares(19).
A partir de la intensidad de la proliferación celular en microscopia óptica, es posible identificar tres categorías de glomerulonefritis:
1. La glomerulonefritis mesangiopática, que se caracteriza por depósitos mesangiales y, eventualmente, por una discreta proliferación mesangial. Este aspecto se observa en el 30 % de los casos.
2. La glomerulonefritis segmentaria y focal, caracterizada por una proliferación celular que afecta solo a un segmento de algunos glomérulos, con una proliferación extracapilar al lado en forma de medialuna segmentaria. Las lesiones evolucionan hacia la fibrosis. Este aspecto se observa en el 45 % de los casos.
3. La glomerulonefritis endocapilar y extracapilar, la forma más grave, en la que una proporción variable de medialunas epiteliales se suma a una proliferación endocapilar difusa.
Evolución
El pronóstico de la enfermedad de Berger parece mejor en los niños que en los adultos. Una de las razones es que puede tratarse de una enfermedad lentamente progresiva; algunos pacientes con enfermedad de Berger que tuvieron los primeros síntomas en la infancia, experimentan una evolución desfavorable en la edad adulta.
Determinados parámetros clínicos guardan relación con un deterioro más frecuente y más veloz de la función renal: mayor edad de comienzo de la enfermedad, proteinuria persistente entre las crisis de hematuria macroscópica, HTA, ausencia de antecedentes de hematuria macroscópica y función renal alterada en el momento del diagnóstico. El pronóstico a largo plazo también se correlaciona con la gravedad de la lesión histológica. Si la biopsia inicial muestra una glomerulonefritis mesangiopática, el pronóstico suele ser favorable. En cambio, la posibilidad de progresión hacia la insuficiencia renal crónica es correlativa con la presencia de semilunas epiteliales o fibrosas(20).
Patogenia
Es desconocida. Se sabe que la nefropatía por IgA es una enfermedad sistémica, como lo demuestra el hecho de que, después de un trasplante renal, los depósitos de IgA vuelven a formarse en el 35 % de los pacientes. Además, cuando un paciente recibe el riñón de un donante que padece nefropatía por IgA asintomática, los depósitos mesangiales de IgA desaparecen enseguida. La enfermedad de Berger es secundaria a complejos inmunitarios que contienen IgA1. La síntesis de IgA1 por linfocitos procedentes de la médula ósea está aumentada. Las IgA1 séricas y las IgA1 identificadas en las biopsias renales de los pacientes tienen un defecto de galactosilación, lo que favorece la formación de agregados de IgA1 y de complejos antígeno-anticuerpo para IgA, así como la fijación de las IgA1 en los receptores de las células mesangiales o en las proteínas de la matriz. La depuración de complejos inmunitarios de IgA por el sistema reticuloendotelial está disminuida. También existen factores genéticos (mayor frecuencia de antígeno HLA B35 y DR4, casos familiares)(21).
Tratamiento
Los niños con anomalías urinarias mínimas no deben recibir tratamiento específico; cerca de una cuarta parte de ellos evoluciona hacia la remisión completa.
Muchos autores indican, sobre todo en el adulto, IECAS para controlar la presión arterial y por su efecto antiproteinúrico. La corticoterapia puede indicarse a pacientes con proteinuria permanente y/o proliferación extracapilar. En varios estudios se ha demostrado que dicho tratamiento (perfusiones de metilprednisolona seguidas de corticoterapia discontinua) puede disminuir la velocidad de progresión de la nefropatía. El ácido eicosapentanoico (EPA) del aceite de pescado se ha ensayado con éxito en algunos estudios realizados en adultos, con disminución de la velocidad de progresión de la enfermedad en pacientes tratados. El tratamiento inmunosupresor debe reservarse para pacientes que no mejoran con IECAS y/o que tienen proteinuria persistente superior a 1 g/d y/o una alteración de la función renal. En enfermos con evolución rápida, se efectúan intercambios plasmáticos de efecto favorable transitorio. La incidencia de insuficiencia renal es significativamente menor en niños sometidos a amigdalectomía(22).
En pacientes que han evolucionado hacia insuficiencia renal terminal, después de trasplante renal, es frecuente la recidiva de los depósitos de IgA en el injerto, pero las modificaciones histológicas de proliferación celular resultan excepcionales.
Glomerulonefritis membranoproliferativa (GNMP)
La GNMP es una enfermedad glomerular crónica. Es poco frecuente en la infancia, pero su importancia radica en que el comienzo puede ser indistinguible de una GNAPE y, en ocasiones, tiene un curso rápidamente progresivo que termina en insuficiencia renal terminal. El curso clínico prolongado del SN, los niveles de C4 descendidos de forma mantenida más allá de 6 semanas y un deterioro rápido de la función renal nos hará realizar una biopsia renal. El estudio histológico muestra un patrón general de daño glomerular caracterizado en la microscopia óptica por un engrosamiento difuso de la membrana basal glomerular con celularidad aumentada. Tradicionalmente, se han clasificado en 3 tipos según la localización de los depósitos: GNMP tipo I, los depósitos de complemento están en el espacio subendotelial; GNMP tipo II, dentro de la lámina densa de la membrana basal glomerular, con una trasformación electrón densa; y GNMP tipo III, subendoteliales y subepiteliales, con laminación y rotura de la membrana basal. Los depósitos en el tipo I y el tipo III contienen inmunoglobulinas (IgM, IgG) y complemento (C3, C4), mientras que en el tipo II o enfermedad de depósitos densos solo contienen C3. Hoy en día, basados en el conocimiento del papel que desempeña el sistema del complemento, nos vemos obligados a cambiar la clasificación y definir, cuando solo encontramos depósitos de C3, la glomerulopatía C3, que tiene un pronóstico malo, con evolución a insuficiencia renal terminal, pero que, por otro lado, tiene una prometedora opción terapéutica con los anticuerpos monoclonales, que inhiben selectivamente el complejo C5b-9, a pesar de que los ensayos clínicos con eculizumab para esta enfermedad no se hayan realizado todavía.
Conclusión
La aproximación a las GNA se realiza de acuerdo a las manifestaciones clínicas, a las pruebas biológicas y, ocasionalmente, a la histología. El cuadro clínico puede ser muy variado, siendo necesario identificar posibles procesos infecciosos recientes, antecedentes familiares, además de los síntomas renales y extrarrenales. Las pruebas de laboratorio que incluyen el examen de orina, el estudio de la función renal y del complemento pueden ser de gran ayuda.
Función del pediatra de Atención Primaria
El síndrome nefrítico se caracteriza por la aparición repentina de edemas e HTA por efecto de la expansión del sector extracelular hacia los espacios vasculares e intersticiales. De forma paralela, existe oliguria con orina turbia. En general, la hematuria es macroscópica y se acompaña de proteinuria, con natriuresis baja. Debe ser considerado en cualquier niño que se presente con síntomas secundarios a sobrecarga de volumen, debiendo realizar un uroanálisis como prueba de diagnóstico inicial.
La sospecha de un síndrome nefrítico obliga a la derivación al Hospital, ya que a menudo se acompaña de una insuficiencia renal moderada, pero en algunos casos puede ser grave y necesitar depuración extrarrenal. La HTA puede complicarse y necesita tratamiento inmediato con hipotensores de acción rápida (diuréticos y antagonistas de calcio).
En la edad pediátrica, las glomerulonefritis agudas habitualmente se presentan en dos contextos clínicos, como una glomerulonefritis aguda postinfecciosa o como una nefritis asociada a una vasculitis por inmunoglobulina A (IgA).
La glomerulonefritis aguda postinfecciosa es una lesión inflamatoria de predominio glomerular y de patogenia inmune desencadenada por gran variedad de gérmenes. Es una enfermedad propia de la edad infanto-juvenil, siendo los niños entre los 4 y los 14 años los más frecuentemente afectados. El prototipo es la glomerulonefritis aguda postestreptocócica y la presentación clínica más característica es el síndrome nefrítico agudo, aunque los casos subclínicos son muy numerosos. El descenso transitorio de C3 sérico es uno de los principales elementos de diagnóstico. Generalmente no está indicada la biopsia para el diagnóstico, reservándose para casos de curso clínico atípico y no hay tratamiento específico.
Las medidas terapéuticas incluyen restricción de líquidos y de sal, tratamiento con diuréticos y erradicación del proceso infeccioso si permanece activo. La evolución a largo plazo en los casos típicos es generalmente favorable, pero no está desprovisto de morbilidad aguda: el síndrome nefrítico agudo requiere ingreso hospitalario y puede cursar con complicaciones graves (edema agudo de pulmón, insuficiencia cardiaca, encefalopatía hipertensiva, necesidad de depuración extrarrenal).
Las glomerulonefritis en el contexto de una vasculitis por IgA pueden requerir un seguimiento ambulatorio conjunto entre el especialista del hospital y el pediatra de Atención Primaria. La nefropatía IgA se presenta inmediatamente después de una infección respiratoria, no suele haber alteración de la función renal y el C3 es normal. La recurrencia de brotes de hematuria, excepcional en la glomerulonefritis postestreptocócica, apoya este cuadro clínico.
Conflicto de intereses
No hay conflicto de interés en la elaboración del manuscrito.
Bibliografía
Los asteriscos reflejan el interés del artículo a juicio de los autores.
1.*** Niaudet P. Poststreptococcal glomerulonephritis. UpToDate. 2020. Stapletton FB, Kim M, editores. Disponible en: http://www.uptodate.com/store.
2.** Rabasco-Ruiz C, Huerta-Arroyo A, Caro-Espada J, Gutiérrez-Martínez E, Praga-Terente M. C3 glomerulopathies. A new perspective on glomerular diseases. Nefrologia. 2013; 33: 164-70.
3.** Rodríguez-Iturbe B, Carr RI, García R, Rabideau D, Rubio L, McIntosh RM. Circulating immune complexes and serum immunoglobulins in acute poststreptococcal glomerulonephritis. Clin Nephrol. 1980; 13: 1-4.
4.** Carapetis JR, Steer AC, Mulholland EK, Weber M. The global burden of group A streptococcal diseases. Lancet Infect Dis. 2005; 5: 685.
5. Ilyas M, Tolaymat A. Changing epidemiology of acute post-streptococcal glomerulonephritis in Northeast Florida: a comparative study. Pediatr Nephrol. 2008; 23: 1101-6.
6. Bingler MA, Ellis D, Moritz ML. Acute poststreptococcal glomerulonephritis in a 14-month-old boy: why is this uncommon? Pediatr Nephrol. 2007; 22: 448-50.
7.** Ahn SY, Ingulli E. Acute poststreptococcal glomerulonephritis: an update. Curr Opin Pediatr. 2008; 20: 157-62.
8. Travis LB, Dodge WF, Beathard GA, Spargo BH, Lorentz WB, Carvajal HF, et al. Acute glomerulonephritis in children. A review of the natural history with emphasis on prognosis. Clin Nephrol. 1973; 1: 169-81.
9. Sjoholm AG. Complement components and complement activation in acute poststreptococcal glomerulonephritis. Int Arch Allergy Appl Immunol. 1979; 58: 274-84.
10. Watanabe T, Yoshizawa N. Recurrence of acute poststreptococcal glomerulonephritis. Pediatr Nephrol. 2001; 16: 598-600.
11. Payne D, Houtman P, Browning M. Acute poststreptococcal glomerulonephritis associated with prolonged hypocomplementaemia. J Clin Pathol. 2008; 61: 1133-5.
12. McCluskey RT, Vassalli P, Gallo G, Baldwin DS. An immunofluorescent study of pathogenic mechanisms in glomerular diseases. N Engl J Med. 1966; 274: 695-701.
13. Yoshizawa N, Suzuki Y, Oshima S, Takeuchi A, Kondo S, Ishida A, et al. Asymptomatic acute poststreptococcal glomerulonephritis following upper respiratory tract infections caused by Group A streptococci. Clin Nephrol. 1996; 46: 296-301.
14.*** Eison TM, Ault BH, Jones DP, Chesney RW, Wyatt RJ. Post-streptococcal acute glomerulonephritis in children: clinical features and pathogenesis. Pediatr Nephrol. 2011; 26: 165-80.
15. Tizard EJ. Henoch-Schönlein purpura. Arch Dis Child. 1999; 80: 380-3.
16.*** Goldstein AR, White RH, Akuse R, Chantler C. Long-term follow-up of childhood Henoch-Schönlein nephritis. Lancet. 1992; 339: 280-2.
17. Lijama K, Ito-Kariya S, Nakamura H, Yoshikawa N. Multiple combined therapy for severe Schönlein-Henoch nephritis in children. Pediatr Nephrol. 1998; 12: 244-8.
18.* Tanaka H, Suzuki K, Nakahata T, Ito E, Waga S. Early treatment with oral immunosuppressants in severe proteinuric purpura nephritis. Pediatr Nephrol. 2003; 18: 347-50.
19.** Levy M, Gonzales-Burchard G, Broyer M, Dommergues JP, Foulard M, Sorez JP, et al. Berger’s disease in children: natural history and outcome. Medicine. 1985; 64: 157-80.
20. Berg U, Bohman SO, Widstam-Attorps U. Renal histological changes in relation to renal function and urinary protein excretion in IgA nephropathy. Arch Dis Child. 1991; 66: 593-7.
21.*** Donadio JV, Grande JP. IgA nephropathy. N Engl J Med. 2002; 347: 738-48.
22.** Floege J. Evidence-based recommendations for immunosuppression in IgA nephropathy: handle with caution. Nephrol Dial Transplant. 2003; 18: 241-5.
Bibliografía recomendada
– Niaudet P. Poststreptococcal glomerulonephritis. UpToDate. 2020. Stapletton FB, Kim M, editores. Disponible en: http://www.uptodate.com/store.
Excelente revisión con especial atención a las manifestaciones clínicas y el diagnóstico de la glomerulonefritis postestreptocócica y el papel de los pediatras en su manejo.
– Ecija Peiró JL. Vázquez Martul M. Glomerulonefritis aguda postinfecciosa. Glomerulonefritis rápidamente progresiva. En: Antón M, Rodríguez LM (coords.). Nefrología Pediátrica: Manual Práctico. Madrid: Editorial Médica Panamericana; 2011. p. 113-8.
Libro de referencia. Revisión del tema con diagnósticos diferenciales entre diferentes formas de glomerulonefritis.
– Donadio JV, Grande JP. IgA nephropathy. N Engl J Med. 2002; 347: 738-48.
Buena revisión de la nefropatía IgA, actualizando conceptos de patogénesis, diagnóstico y pronóstico.
– Fernández Maseda MA, Romero Sala FJ. Glomerulonefritis aguda postinfecciosa. Protoc diagn ter pediatr. 2014; 1: 303-14.
Excelente protocolo y diagnóstico diferencial.
| Caso clínico: glomerulonefritis aguda postinfecciosa |
|
Juan es un niño de 10 años, que es llevado al servicio de urgencias por sus padres, por haber presentado dos episodios de convulsiones tónico-clónicas unas horas antes de su llegada al hospital. Refiere cefalea intensa asociada a fotofobia de 5 días de evolución. Presenta vómitos en escopetazo no relacionados con las comidas. No presenta fiebre ni signos respiratorios. Ha tomado en casa infusiones (medicina tradicional) y paracetamol sin mejoría de la cefalea. Antecedentes Presenta buen desarrollo psicomotor. Es el menor de tres hijos, nacido a término en un hospital, con llanto espontáneo al nacer. Presenta calendario vacunal correcto (DTP polio, hepatitis B, triple vírica) y nunca antes había tenido convulsiones. Antecedentes familiares sin interés por parte de sus padres. Su abuela materna es diabética e hipertensa. Es su primer ingreso hospitalario. Las serologías frente al virus de la inmunodeficiencia humana (VIH), hepatitis C y sífilis son negativas. Presentación del caso A su llegada, Juan está somnoliento, presenta un peso de 34,7 kg, una altura de 143 cm (percentil 75), pulso de 83 lpm, temperatura axilar de 36,8ºC, frecuencia respiratoria de 24 rpm, saturación de oxígeno del 96 %, glucemia de 108 mg/dL y presión arterial de 141/106 mmHg. Presenta un leve edema de los párpados con pupilas isocóricas y normorreactivas a la luz. Ausencia de caries dental y leve edema maleolar. Su auscultación cardiopulmonar es normal. No presenta ascitis ni refiere dolor a la exploración del abdomen. No presenta edema escrotal. Durante su evaluación presenta movimientos tónico-clónicos. No presenta lesiones cutáneas ni los padres refieren oliguria. En la tira reactiva de orina se observa sangre 3+, proteínas 1+, pH 6, densidad de 1,030, cuerpos cetónicos 2+ y resto sin interés. Retomando la anamnesis, ha presentado amigdalitis tratada con betalactámicos durante los 12 días previos al inicio del cuadro. Sospecha clínica de encefalopatía hipertensiva secundaria a una GNAPI/HTA secundaria a estudio. Tratamiento en la unidad de cuidados intensivos En las pruebas de laboratorio se observa: hemoglobina: 120 g/l; leucocitos: 14.300/µl; neutrófilos: 89 %; plaquetas: 145.000; grupo sanguíneo: B+; serología de sífilis y VIH: negativas; natremia: 137 mEq/l; potasemia: 3,1 mEq/l; cloro: 96 mEq/l; creatinina sérica: 0,70 mg/dl; proteínas totales séricas: 67 g/l; albúmina sérica: 40 g/l (37-45 g/l). En orina: 200-250 hematíes, 10-12 leucocitos. Tratamiento: se administra furosemida a dosis de 1 mg/kg cada 8 horas con monitorización estricta de los signos vitales y de la diuresis. Alimentación parenteral estricta con restricción hidrosódica. Aproximadamente a las 10 horas de su ingreso, presenta un nuevo episodio de convulsiones tónico-clónicas precedidas por un vómito en escopetazo. Presenta una presión arterial de 160/130 mmHg con trastornos visuales. No se observa focalidad neurológica y la auscultación cardiopulmonar es normal. Se administra labetalol intravenoso continuo durante 48 horas asociado a furosemida, que permite un mejor control de la presión arterial. Al 3er día del ingreso se observa una reanudación de la diuresis con desaparición progresiva de los edemas, pero con persistencia de la hematuria macroscópica. Al 4° día del ingreso, su presión arterial sigue siendo superior al percentil 99, pero ya no presenta convulsiones. La ecografía renal al 4° día muestra una desdiferenciación corticomedular bilateral con un tamaño renal y Doppler normales. Se añade al tratamiento nifedipino, y a las 24 horas, es decir, al 5° día del ingreso, se suspende el labetalol. En conjunto, presenta una evolución clínica satisfactoria. Juan es dado de alta sin necesidad de tratamiento con diuréticos o hipotensores. A las 24 horas del alta los resultados del estudio del complemento son: C3: 0,64 g/l (0,75-1,40 g/l) y C4: 0,08 g/l (0,10-0,34). ASLO: 800 UI/l. Al día siguiente a su ingreso se realizó un fondo de ojo que resultó normal. Se realiza un seguimiento ambulatorio regular con desaparición de la hematuria microscópica a los 5 meses y normalización del complemento a los 2 meses del alta hospitalaria. |

 Hypercalciuria and hypocitraturia. The concept of prelithiasis in Pediatrics
Hypercalciuria and hypocitraturia. The concept of prelithiasis in Pediatrics