 |
| Temas de FC |
P.M. Rubio Aparicio, B. Rosich del Cacho
Unidad de Hemato-Oncología Pediátrica. Hospital Universitario La Paz
| Resumen
La neoplasia más importante derivada de la cresta neural es el neuroblastoma, que representa el tumor sólido extracraneal más frecuente en niños (10% de la incidencia de cáncer pediátrico y 15% de la mortalidad). |
| Abstract
The main neural crest derived neoplasia is neuroblastoma, representing the most frequent solid extracranial tumor in children (with an incidence of 10% of pediatric cancer and 15% of mortality). |
Palabras clave: Neuroblastoma; NMYC; Tumor abdominal; Inmunoterapia; Feocromocitoma; Paraganglioma
Key words: Neuroblastoma; MYCN; Abdominal tumor; Immunotherapy; Pheochromocytoma; Paraganglioma
Pediatr Integral 2016; XX (7): 434-446
Tumores de la cresta neural
Introducción
El neuroblastoma es, por su frecuencia y agresividad, el más importante de los tumores derivados de la cresta neural.
Se trata del tumor maligno más frecuente en el primer año de vida, y del primer tumor sólido extracraneal en la edad pediátrica (0 a 14 años). Representa el 10% de la incidencia del cáncer infantil (13,9 casos por 100.000 niños en España) y el 15% de la mortalidad(1,2). Mientras la supervivencia a 5 años del diagnóstico supera el 90% en pacientes menores de un año, en los casos de riesgo alto, apenas llega al 60%(3). Reducir el tratamiento en pacientes favorables e incrementar la supervivencia en los desfavorables es el reto actual. Para ello, están en marcha técnicas cada vez más avanzadas de diagnóstico genético y molecular, así como ensayos clínicos con terapias dirigidas con el objetivo de personalizar el tratamiento desde el diagnóstico.
Otros tumores derivados de la cresta neural, como el feocromocitoma o el paraganglioma, serán también revisados brevemente.
Fisiopatología y oncogénesis
Los tumores neuroblásticos derivan de los precursores simpaticoadrenales procedentes de la cresta neural, cuando son incapaces de diferenciarse a tejido neural maduro y resisten a las señales de apoptosis.
La cresta neural es un conjunto de precursores multipotenciales que surge bajo el ectodermo en la región dorsal del tubo neural, cuando este se cierra. De ella, se origina el sistema nervioso periférico, el sistema nervioso entérico y la médula adrenal, entre otros tejidos.
En concreto, las células de linaje simpaticoadrenal generan los ganglios simpáticos y la médula adrenal, y se sospecha que podrían ser la estirpe causante del neuroblastoma.
Durante el desarrollo embrionario normal, las células de la cresta neural sufren un proceso conocido como “transición epitelio mesenquimal” (EMT), que les proporciona la capacidad de migrar y desplazarse. Los precursores simpaticoadrenales se dirigen ventralmente hacia la aorta dorsal, para posteriormente, diferenciarse en células cromafines adrenales, células catecolaminérgicas adrenales o ganglios simpáticos (Fig. 1).
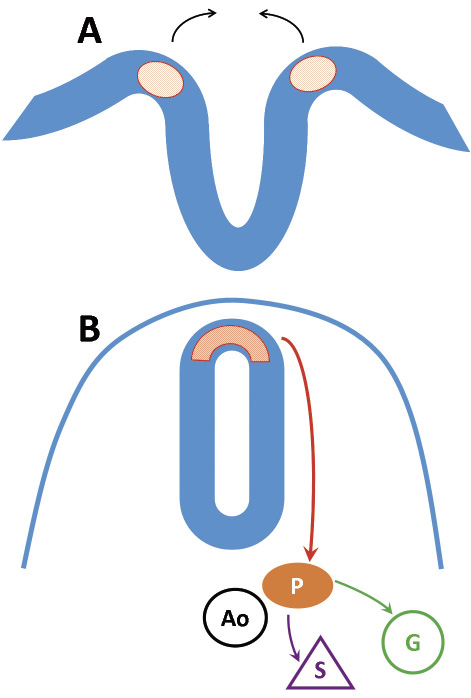
Figura 1. Representación esquemática de la migración y diferenciación desde la cresta neural. A: cierre del tubo neural. B: las células de la cresta neural (punteado rojo) sufren una transición epitelio-mesenquimal (EMT), se delaminan del tubo neural y migran ventralmente hacia la aorta dorsal (Ao) para formar los progenitores simpaticoadrenales (P). Posteriormente, continúa su migración y diferenciación a la médula suprarrenal (S) o a los ganglios simpáticos (G). Adaptado de: Jiang M, et al. Curr Top Dev Biol 2011.
Durante todo el desarrollo del sistema nervioso periférico existe un equilibrio entre la proliferación, migración, diferenciación y muerte celular. Un error en cualquiera de estos procesos puede derivar en la transformación celular tumoral.
Así, el oncogén NMYC, necesario para la supervivencia de los neuroblastos a lo largo de todas estas fases, se encuentra amplificado en los neuroblastomas de riesgo alto. De hecho, los mecanismos de metástasis del neuroblastoma son equivalentes a los que permiten la migración durante la EMT.
Los receptores para tropomiosina (Trk), también juegan un papel importante. Los neuroblastomas que expresan TrkA son sensibles al factor de crecimiento neural (NGF), sin el cual entran en apoptosis, y en cuya presencia maduran a formas benignas. En cambio, si expresan TrkB, se produce un estímulo autocrino de la proliferación, ya que su ligando, el factor neurotrófico derivado del cerebro (BDNF), es sintetizado por los propios neuroblastos. Existe una asociación entre la expresión de TrkB y la amplificación de NMYC(4).
Clínica
La heterogeneidad es una de las marcas distintivas del neuroblastoma, que puede aparecer en cualquier punto del sistema nervioso simpático, por lo que la localización e intensidad de los síntomas son muy variables(5).
Dos tercios de los casos aparecen en el abdomen, la mayoría suprarrenales, o como masas retroperitoneales. El resto se distribuyen en región cervical, torácica y pélvica. Habitualmente, se localizan en regiones posteriores, a menudo paravertebrales.
El neuroblastoma puede ser un hallazgo casual en un niño asintomático, lo que sucede cada vez con más frecuencia, debido al aumento de las ecografías de control, especialmente en lactantes.
En los tumores localizados, los síntomas dependerán de su ubicación, su tamaño y de la compresión de estructuras vecinas. Es típico el síndrome de Claude-Bernard-Horner (ptosis, miosis y enoftalmos) en masas cervicales.
La compresión espinal puede producir déficits motores y sensitivos y constituye una emergencia oncológica que requiere laminectomía descompresiva y/o quimioterapia de urgencia.
Puede existir linfedema e hidrocele en varones; es raro observar síntomas de obstrucción intestinal o urinaria.
Las metástasis pueden ser sintomáticas. Los lugares más frecuentes son: el hueso, con especial tropismo por los huesos orbitarios, dando lugar a proptosis o equimosis con “ojos de mapache” (en otras localizaciones, puede producirse dolor y/o impotencia funcional, raramente fractura); la médula ósea, pudiendo haber signos de hipoplasia medular; y el hígado, donde, sobre todo, produce efecto masa sin afectación de la funcionalidad hepática. Puede haber metástasis cutáneas en forma de nódulos violáceos.
Los lactantes tienen un pronóstico muy favorable y, sin embargo, pueden presentar al debut una situación de riesgo vital, debido a la desproporción entre el tamaño tumoral y el del paciente, especialmente en casos con metástasis hepáticas.
La producción tumoral de catecolaminas y la compresión del hilio renal pueden producir hipertensión arterial. En ocasiones, el tumor también producirá péptido intestinal vasoactivo (VIP), con diarrea que no mejora con el tratamiento del neuroblastoma.
El síndrome de opsoclono mioclono (OMS) es un cuadro de naturaleza autoinmune por reactividad cruzada entre antígenos asociados al neuroblastoma y estructuras del sistema nervioso central.
Aunque solo el 2% de los neuroblastomas asocian un OMS, se estima que se hallará un neuroblastoma como causa subyacente en aproximadamente la mitad de los OMS.
El OMS se caracteriza por irritabilidad, temblor, movimientos oculares anómalos y pérdida de hitos psicomotores (marcha, bipedestación y sedestación). Se asocia a neuroblastomas de pronóstico favorable. El tratamiento se basa en la administración de dexametasona y/o gammaglobulina intravenosa. Dos tercios de los pacientes presentan secuelas en el aprendizaje, la marcha y/o el lenguaje, y son frecuentes las recurrencias, aunque esté resuelto el neuroblastoma, de cuyo curso es independiente. Actualmente, se está investigando de manera prospectiva la administración de ciclofosfamida en casos con mala respuesta a dexametasona, y del anticuerpo monoclonal anti-CD20 Rituximab en casos refractarios(6).
Diagnóstico
Además de la radiología convencional y de la obtención de muestra de tumor para estudio histológico y genético, forman parte esencial del estudio diagnóstico inicial: la determinación de catecolaminas urinarias, el estudio de extensión gammagráfico y el estudio de médula ósea.
La prueba diagnóstica inicial de elección es la ecografía, por su accesibilidad e inocuidad y su capacidad de elevar una sospecha diagnóstica y conducir al resto de estudios. La prueba anatómica de elección es la resonancia magnética nuclear (RMN), reemplazando a la tomografía computerizada (TC), debido a la ausencia de radiación y a la buena definición de la afectación espinal (Fig. 2).
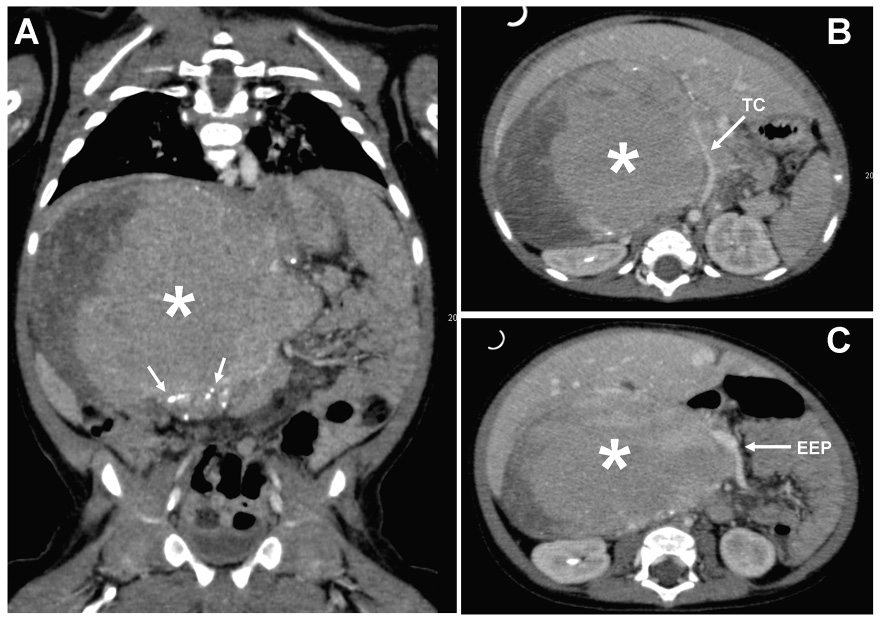
Figura 2. Gran masa (102 x 124 mm) abdominal derecha (*) que cruza la línea media desplazando y englobando estructuras vasculares. A: corte coronal. Las flechas señalan áreas de calcificación. B y C: cortes axiales. TC: tronco celíaco. EEP: eje espleno-portal. Cortesía de la Dra. Pérez Vigara.
La prueba de elección para el estudio de extensión es la gammagrafía con metayodobencilguanidina (MIBG) marcada con yodo radioactivo. Se trata de un análogo de noradrenalina que es captado a nivel presináptico por el transportador de noradrenalina (NAT).
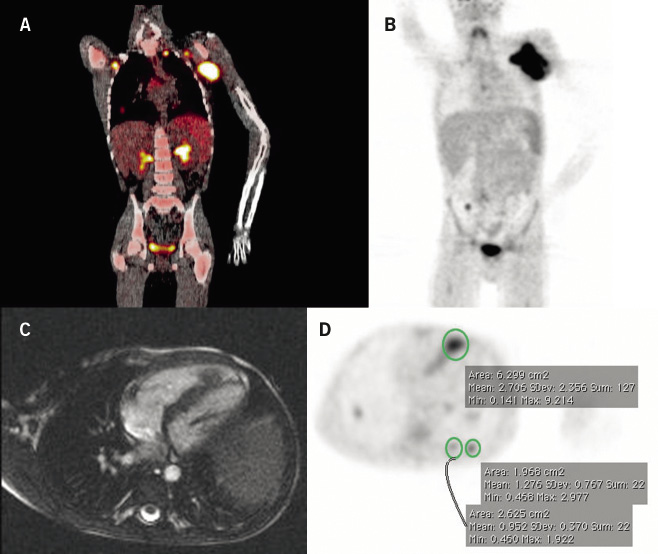
Es muy sensible y específica; más de 90% de los neuroblastomas tienen avidez por MIBG. Se utiliza el isótopo 123I, trazador ideal para diagnóstico con vida media corta, baja irradiación, y que permite realizar reconstrucciones tridimensionales mediante SPECT-TC (Fig. 3).
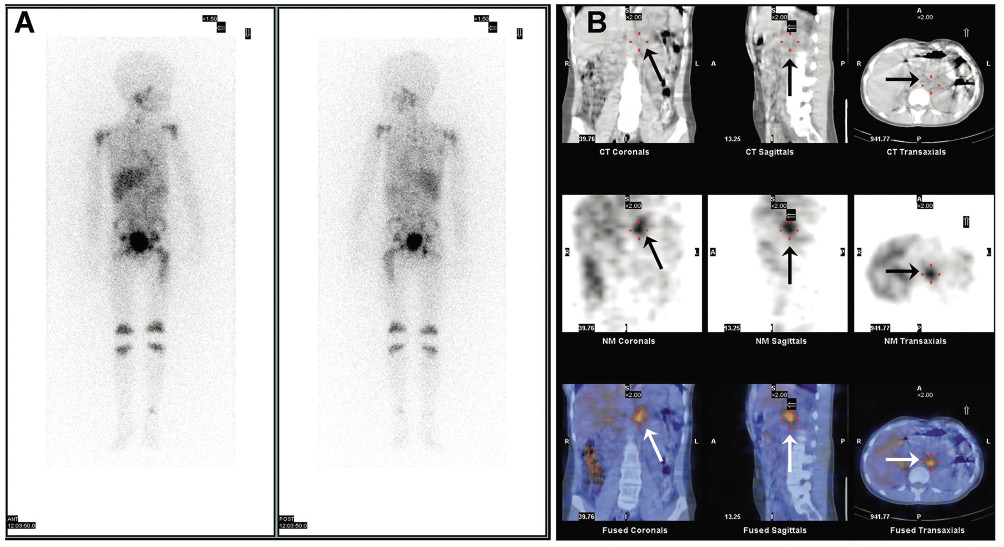
Figura 3. Estudios de Medicina Nuclear. A: gammagrafía con 123I-MIBG en la que, además del depósito fisiológico en hígado, vejiga y glándulas salivales, se observan captaciones óseas patológicas (calota, tercio proximal de ambos húmeros, raquis, huesos pélvicos, ambos fémures y rodillas). B: integración de TC y 123I-MIBG-SPECT (SPECT-TC), observándose una lesión paraespinal izquierda (flechas). Cortesía de la Dra. Rodado.
Como inconvenientes, en casos de masas grandes o muy ávidas, el tumor primario puede absorber todo el trazador y, por tanto, infraestimarse las metástasis; además, existe aproximadamente un 10% de neuroblastomas no captantes de MIBG.
Debido a que la interpretación de la MIBG es subjetiva, para minimizar las diferencias interobservador se han propuesto diversas escalas semicuantitativas (CURIE, SIOPEN), habiéndose encontrado correlación entre los valores absolutos y relativos (es decir, la diferencia entre el valor diagnóstico y postquimioterapia) y la supervivencia(7).
La gammagrafía ósea con 99Tc era la prueba de extensión vigente hasta la implantación de la MIBG, pero presenta falsos negativos (lesiones incipientes sin aumento de resorción ósea) y positivos (por aumento de remodelado en lesiones resueltas).
La tomografía por emisión de positrones de 18 fluorodeoxiglucosa combinada con TC (18FdG-PET-TC), se ha propuesto como alternativa a la MIBG, con buenos resultados.
La PET-TC ofrece la ventaja de que no existe saturación en casos con tumores primarios de gran tamaño y avidez, y parece detectar mejor la enfermedad en tejidos blandos; mientras que podría ser algo inferior a la hora de evaluar la afectación ósea. Otros marcadores, como 124I se están estudiando actualmente, en diversos ensayos clínicos.
La realización de aspirado-biopsia bilateral de médula ósea, es una parte esencial del estudio de extensión. Además del estudio histológico, se está evaluando el valor de la detección por PCR de marcadores (doblecortina, tirosín-hidroxilasa y PHOX2B) en sangre periférica y médula ósea(8).
El objetivo es que estos marcadores, una vez validados, permitan incorporar estudios de enfermedad mínima residual a la rutina diagnóstica y de evaluación de respuesta.
En cuanto a los estudios analíticos, tanto hemograma como bioquímica suelen ser normales. Se puede observar hiperferritinemia y elevación de lactato deshidrogenasa (LDH). Es característica la elevación de la enzima neural enolasa neuroespecífica (NSE). En caso de infiltración medular masiva, puede haber citopenias, aunque no es un hallazgo habitual.
Al ser un tumor productor de catecolaminas, puede haber elevación en orina de estas y sus metabolitos. La detección de dopamina, ácido homovanílico y vanilmandélico en orina forma parte del estudio diagnóstico y del seguimiento de la enfermedad(9).
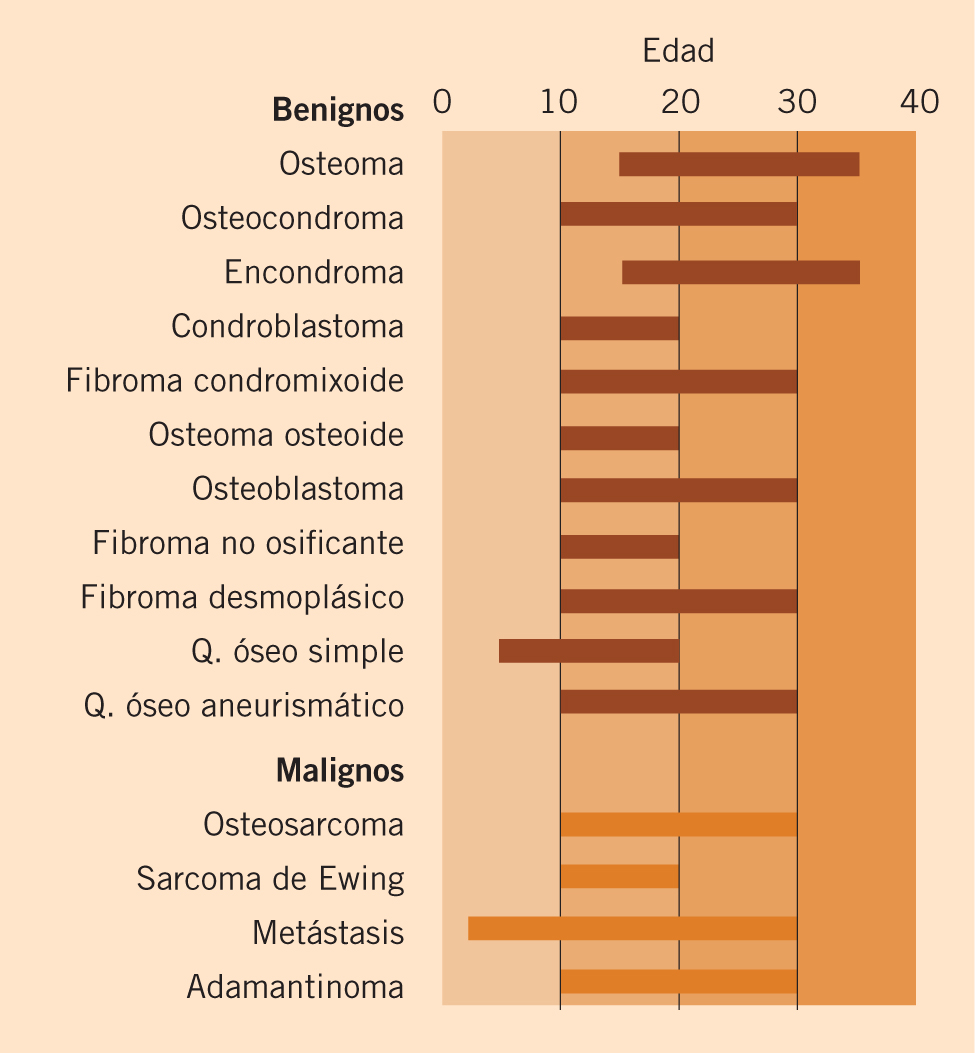
Por último, el diagnóstico confirmatorio es anatomopatológico, mediante el estudio histológico y genético del tumor. La clasificación histopatológica vigente es la establecida por Shimada en 1999, que discierne entre casos favorables y desfavorables en función de la edad, el grado de diferenciación y la relación entre mitosis y cariorrexis (Tabla I)(10).

En cuanto a los estudios genéticos, la hibridación fluorescente in situ (FISH) para detectar amplificación de NMYC y deleción de 11q, está siendo reemplazada por el uso de matrices de polimorfismos de nucleótido simple (SNPs arrays), mucho más informativas (Fig. 4).

Figura 4. A: histología de neuroblastoma pobremente diferenciado. Se observan neuroblastos en un estroma finamente fibrilar con focos de necrosis y calcificación. B: FISH que muestra amplificación de NMYC (rojo, marcado con asteriscos) frente al gen control LAF (verde, marcado con flechas). C: SNPs array, en el que se observa pérdida de 1p y 10q y ganancia de 17q, así como amplificación de NMYC en 2p (NMA). D: detalle del cromosoma 2, en el que se observa la amplificación de NMYC (2p24). Cortesía de los Drs. Font de Mora, Regojo y Berlanga.
Las técnicas utilizadas actualmente en investigación no se limitan al ADN, sino que abarcan desde los moduladores de su expresión (epigénetica) hasta sus productos (ARNoma, microARN, proteoma). Se espera que la información cada vez más completa y compleja sobre la biología tumoral proporcione nuevas herramientas diagnósticas y terapéuticas (Fig. 4)(11).
Genética
La heterogeneidad clínica del neuroblastoma tiene una base biológica, en función de la aparición en el desarrollo tumoral de alteraciones cromosómicas numéricas (de buen pronóstico) o segmentarias (desfavorables).
Los casos con comportamiento favorable tienen un contenido de ADN hiperdiploide (a menudo, triploide) a expensas de ganancia de cromosomas enteros, sin reordenamientos citogenéticos. Este perfil se conoce como NCA (Numerical Chromosomal Aberrations). En cambio, en los desfavorables se detecta un perfil SCA (Segmental Chromosomal Aberrations), caracterizado por la aparición de alteraciones cromosómicas segmentarias, con pérdidas y ganancias parciales de cromosomas (17q+, 1p-) y un contenido en ADN variable, desde casi-diploide a casi-tetraploide.
Los tumores NCA expresan TrkA y, además de a NGF, son sensibles a tratamientos diferenciadores, como el ácido 13-cis retinoico (13cisRA): tienden a madurar a formas benignas o a la apoptosis de manera espontánea.
En cambio, los casos con perfil SCA no maduran ni regresan espontáneamente, tienen un comportamiento agresivo y tendencia a la recaída. Expresan receptores TrkB. Hay dos perfiles diferenciados, mutuamente excluyentes: los pacientes con amplificación de NMYC, principal factor de mal pronóstico en los protocolos vigentes, y que condiciona una baja supervivencia a corto plazo y una elevada tasa de recaídas precoces; y los casos con deleción del brazo largo del cromosoma 11 (11q-), con un curso insidioso marcado por sucesivas recaídas tras respuesta al tratamiento, que implica una supervivencia más prolongada, pero con una tasa de mortalidad similar, si no superior, a los casos con amplificación de NMYC (Fig. 5)(12,13).
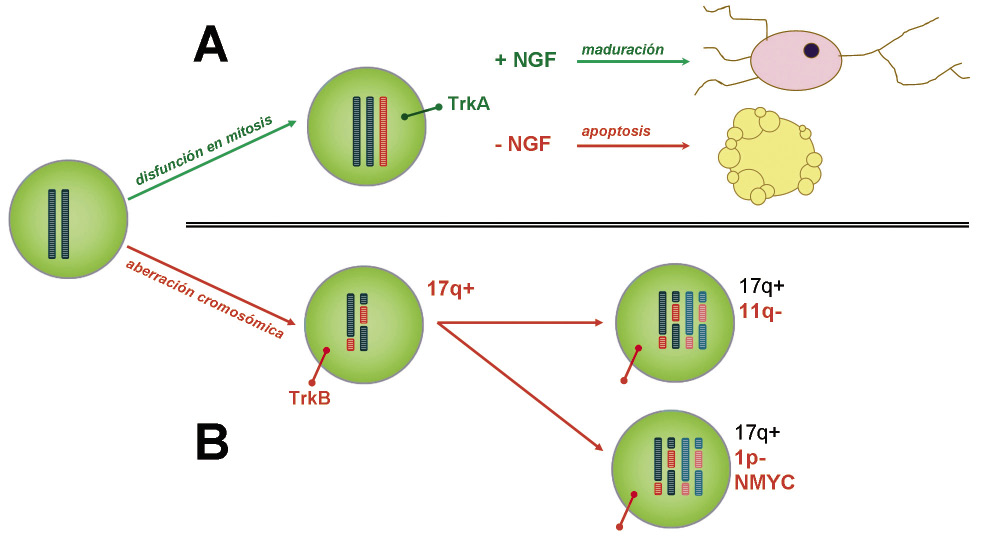
Figura 5. Oncogénesis del neuroblastoma. En los casos favorables (A), se produce una disfunción en la mitosis que genera neuroblastos con un número anormal de cromosomas estructuralmente normales (perfil NCA). Son células triploides que expresan receptores TrkA. En presencia de agentes diferenciadores (+ NGF), maduran a tejido ganglionar, y en su ausencia (-NGF), entran en apoptosis. En los casos desfavorables (B99, se producen alteraciones cromosómicas estructurales (perfil SCA). Son células diploides o tetraploides que acumulan ganancias y pérdidas parciales de cromosomas. Expresan receptores TrkB que favorecen la proliferación. Se distinguen dos grupos mutuamente excluyentes: los casos con deleción de 11q no suelen tener amplificado NMYC, y viceversa. TrkA/B: kinasa del receptor de tropomiosina. NGF: factor de crecimiento neural. Adaptado de: Brodeur GM. Nat Rev Ca 2003.
Dentro de los nuevos marcadores actualmente en estudio, destaca la kinasa del linfoma anaplásico (ALK). Se trata de una tirosín kinasa cuya mutación/sobreexpresión se asocia a las formas familiares de neuroblastoma (menos del 5% de los casos) y al 10% de los neuroblastomas esporádicos. Es el más prominente de los nuevos marcadores en estudio, por su implicación pronóstica y su posible utilidad como diana terapéutica(4,11,14).
Estadificación y factores pronósticos
El sistema actual de estadificación, vigente desde 2008, se basa en la extensión de la enfermedad, la edad, el tipo histológico y algunos rasgos biológicos del tumor. Se establecen cuatro estadios: dos localizados (L1 y L2) y dos metastáticos (M y Ms) (Tabla II)(15).

Los tumores localizados (L) se dividen en dos grupos (L1 y L2), en función de si son operables o no al diagnóstico sin riesgo de complicaciones. Para ello, existen unos criterios estandarizados conocidos como Factores de Riesgo Definidos por Imagen (IDRF). Los pacientes que no presentan IDRF (estadio L1) se operan de inicio; los pacientes con uno o más IDRF (estadio L2) tienen contraindicada la cirugía (Tabla III).

Los tumores metastásicos se clasifican como estadio M y son de riesgo alto, exceptuando un grupo denominado Ms, formado por niños pequeños (hasta 18 meses de edad) y con metástasis limitadas a hígado, piel y/o médula ósea. Los pacientes estadio Ms tienen paradójicamente un pronóstico excelente y se consideran de riesgo bajo.
La edad es un factor pronóstico clave, con una clara diferencia de supervivencia entre niños menores y mayores de un año, aunque el punto de corte podría situarse más cercano a los 18 meses de edad(3,16).
La histología permite discernir entre pacientes de riesgo bajo-intermedio, siendo el pronóstico más favorable cuanto más maduro sea el tumor.
Aunque la estadificación actual solo incluye como marcadores pronósticos genéticos la amplificación de NMYC, la deleción de 11q y la ploidía (diploide o hiperploide), de manera práctica se están utilizando los perfiles NCA/SCA a la hora de elegir tratamiento, especialmente en niños menores de 18 meses. Se espera que el próximo sistema de estadificación dé mucho mayor peso a los factores biológicos (Tabla IV)(11).

Tratamiento
Además de ser susceptible a extirpación quirúrgica, el neuroblastoma es quimio y radiosensible, y uno de los pocos tumores sólidos en los que la quimioterapia mieloablativa con rescate autólogo de progenitores hematopoyéticos (ATPH) ha demostrado efectividad. La inmunoterapia es la incorporación más novedosa al tratamiento de primera línea.
Existen tres grandes objetivos: estandarizar el tratamiento de primera línea y en recaída en protocolos multicéntricos y plurinacionales, disminuir la toxicidad del tratamiento en pacientes de riesgo bajo, y aumentar la supervivencia en los de riesgo alto.
Estadio L1
Los tumores localizados sin factores de riesgo quirúrgico (IDRF negativos) son subsidiarios de extirpación quirúrgica, como único tratamiento.
Debido a su excelente pronóstico, se propugna no operar, sino observar estrechamente a los lactantes menores de 3 meses de edad con masas adrenales pequeñas (menores de 5 cm). Solo se realizaría cirugía, en caso de observarse crecimiento de la masa, o su persistencia cumplido el año de vida.
Si se detecta amplificación de NMYC en la pieza quirúrgica, la extirpación no se considera tratamiento suficiente, y se recomienda consolidación con quimio y radioterapia.
Grupos de riesgo bajo e intermedio
Los lactantes (hasta 18 meses) con enfermedad localizada con IDRF positivos (L2), o metástasis limitadas a hígado, médula ósea y/o piel (Ms), tienen un pronóstico muy favorable con supervivencia superior al 90% sin tratamiento citotóxico.
Las estrategias actuales buscan minimizar el tratamiento quimioterápico de estos pacientes. Las indicaciones de quimioterapia se limitan a la presencia de rasgos biológicos adversos (perfil SCA) o de síntomas amenazantes para la vida que hagan imprescindible la reducción rápida de la masa tumoral (Tabla V).

Existe un grupo de pacientes que puede considerarse de riesgo intermedio, al presentar a la vez, características favorables y desfavorables: estadios L1 extirpados, con amplificación de NMYC; estadios L2, en niños mayores de 18 meses; y lactantes de hasta 12 meses de edad, con estadio M. Estos pacientes se tratan con quimioterapia neodayuvante de intensidad variable, seguida de cirugía si procede y, en algunos casos, con radioterapia y 13cisRA.
Grupos de riesgo alto
Los pacientes con NMYC amplificado, y los estadios M en mayores de 12 meses, comprenden un grupo desfavorable con elevada tasa de recaídas y mortalidad. Su tratamiento se compone de tres fases: inducción de remisión (quimioterapia y cirugía), consolidación (ATPH y radioterapia) y mantenimiento (inmunoterapia y 13cisRA).
Como se mencionó previamente, los pacientes L1 con NMYC amplificado son una excepción y no reciben tratamiento de riesgo alto, sino intermedio.
La fase de inducción dura 3-4 meses y busca eliminar toda enfermedad detectable. Se inicia con poliquimioterapia para eliminar las metástasis (Tabla VI).

Al finalizar la quimioterapia, si la respuesta de las metástasis ha sido adecuada (desaparición completa; o disminución del 50% con persistencia de un máximo de tres lesiones) se procede a la extirpación quirúrgica completa del tumor primario.
Los pacientes que no han presentado respuesta metastásica adecuada tras la inducción, pasan a recibir tratamiento de segunda línea (habitualmente, ensayos clínicos fase II) como casos refractarios.
Los lactantes entre 12 y 18 meses con estadio M, si tienen una biología favorable (NCA y NMYC no amplificado) acaban aquí el tratamiento. El resto de pacientes pasa a consolidación y mantenimiento.
La fase de consolidación dura 4 meses y comprende un tratamiento sistémico, mediante la administración de quimioterapia mieloablativa con busulfan y melfalan, seguida de rescate autólogo de progenitores hematopoyéticos (ATPH); y uno local, mediante radioterapia sobre el lecho tumoral. La irradiación de lesiones óseas prominentes al diagnóstico podría reducir el porcentaje de recaídas, pero también ocasionar secuelas. Se está estudiando modular la intensidad de la radioterapia en función de la respuesta a la fase de inducción (Mark Gaze, comunicación personal, SIOPEN AGM 2015).
Por último, la fase de mantenimiento dura 9 meses y se basa en la administración de inmunoterapia, asociando el anticuerpo monoclonal antiGD2 dinutuximab con inmunoestimuladores (interleukina-2, GM-CSF), y con el agente diferenciador 13cisRA. Esta fase es la más novedosa y está basada en la relativa especificidad de dinutuximab, ya que su antígeno diana, el disialogangliósido GD2, se encuentra casi en exclusividad en la membrana de los tejidos neurales embrionarios. Su eficacia se acompaña de toxicidad importante, especialmente dolor neuropático y reacciones alérgicas, que han obligado a adoptar estrategias de infusión prolongada para reducir efectos adversos(17).
A pesar de la intensidad, duración y toxicidad de este tratamiento, la supervivencia libre de enfermedad (SLE) a 3 años del diagnóstico de un neuroblastoma de riesgo alto, se sitúa en el 63%.
Existe un dato esperanzador, debido a que en los pacientes que han podido recibir todo el tratamiento sin reducción de dosis por toxicidad, la SLE a 3 años sube hasta el 77% (R. Ladenstein, comunicación personal, SIOPEN AGM 2015).
Recaídas y refractarios
La tasa de recaídas en neuroblastoma es elevada, pudiendo llegar al 50% en los pacientes de riesgo alto.
Las estrategias actuales pasan por el uso de quimioterapia con combinaciones de: temozolamida con irino/topotecan; la radioterapia dirigida con 131I-MIBG; la quimioterapia intensiva con autotrasplante en tándem; y la terapia antiangiogénica (vorinostat). Existe un importante esfuerzo para establecer protocolos estandarizados para estos pacientes. La aparición de nuevas terapias abre también nuevas posibilidades en forma de ensayos clínicos. Aun así, la supervivencia a largo plazo tras recaída no llega al 10%(11).
Tratamiento de urgencia
Existen dos casos que requieren tratamiento urgente: la compresión espinal sintomática y los síntomas amenazantes para la vida en neonatos (habitualmente, en pacientes estadio Ms con metástasis hepáticas masivas).
En el primero de los casos, puede estar indicada la laminectomía descompresiva. En ambos, puede administrarse quimioterapia incluso antes de obtener muestra de tumor, habitualmente, asociando etopósido y carboplatino. Es importante ser cauteloso con el tratamiento quimioterápico en neonatos por su elevada toxicidad.
Nuevas terapias
Existen múltiples aproximaciones terapéuticas que se pueden resumir en tres grupos: la radioterapia dirigida, las moléculas pequeñas inhibidoras y la inmunoterapia (Fig. 6).
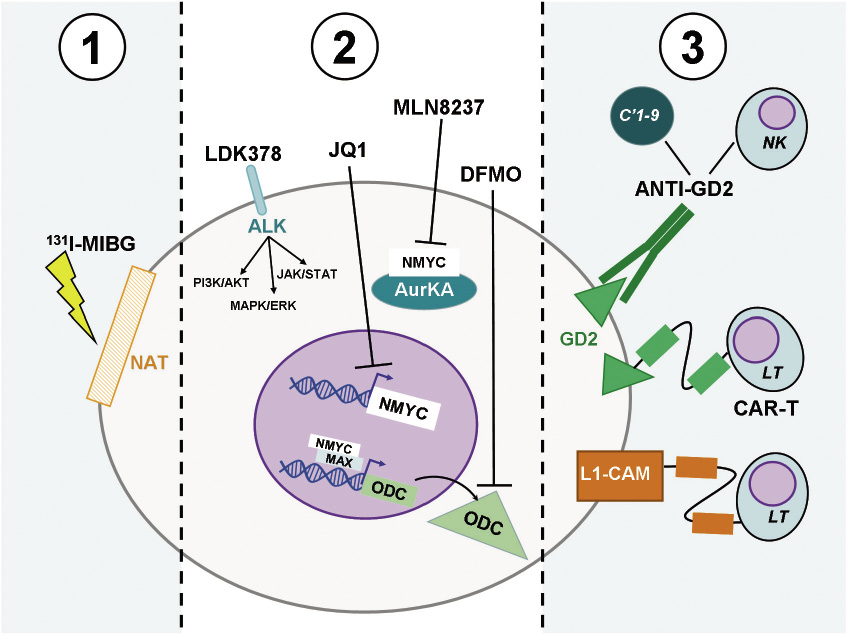
Figura 6. Nuevas dianas terapéuticas en neuroblastoma. 1: radioterapia dirigida mediante la administración de 131I-MIBG, captada por el transportador de noradrenalina (NAT). 2:inhibidores de molécula pequeña: LDK378: inhibición de la kinasa de linfoma anaplásico (ALK) y sus vías de señalización; JQ1: inhibición de bromodominios para evitar la transcripción de NMYC; MLN8237: inhibición de Aurora Kinasa A (AurKA) para desestabilizar NMYC; y DFMO (difluorometilornitina): bloqueo de ornitín decarboxilasa (ODC), factor limitante de la síntesis de poliaminas. 3: inmunoterapia: anticuerpos quiméricos antidisialogangliósido (GD2) que median destrucción inmune vía complemento y citotoxicidad derivada de anticuerpo (ADCC); y linfocitos T con receptores de antígeno quimérico (CAR-T) dirigidos contra GD2 y la molécula de adhesión celular L1 (L1-CAM). PI3K/AKT: fosfatidil inositol 3-kinasa. MAPK/ERK: proteín kinasa activada por mitógenos. JAK/STAT: Janus kinasa/transductor de señal y activador de la transcripción. Adaptado de: Pinto NR. JCO 2015.
La administración de altas dosis de 131I-MIBG, captada vía NAT, es una opción eficaz sobre la que hay amplia experiencia. Se puede administrar en monoterapia o combinada con sensibilizantes, como irinotecan o topotecan. Debido a su toxicidad medular, suele acompañarse de rescate autólogo de progenitores hematopoyéticos.
Existen diversas dianas para la inhibición mediante moléculas pequeñas. La más relevante es ALK. El inhibidor de primera generación crizotinib no demostró eficacia; se está evaluando la eficacia de moléculas de segunda generación como LDK378 (ceritinib).
La inhibición de Aurora Kinasa A, una serín-treonín kinasa citoplasmática que estabiliza NMYC, la inhibición de la propia transcripción de NMYC vía proteínas BET (Bromodomain and Extraterminal Motif) o de la síntesis de poliaminas mediante el bloqueo de ornitindecarboxilasa (ODC) con difluorometinornitina (DFMO), son otras aproximaciones de terapia dirigida.
Además del anticuerpo monoclonal antiGD2, la aproximación inmunoterapéutica más novedosa es la técnica CAR (Chimeric Antigen Receptor), que consiste en la incorporación ex vivo de receptores contra antígenos específicos del tumor (en este caso, GD2 y L1-CAM) a linfocitos T del paciente. Otras aproximaciones, consideran la potenciación de la inmunovigilancia ejercida por los linfocitos NK, mediante su expansión o mediante suplementos como arabinoxilano(19).
Feocromocitoma y paraganglioma
Se trata de tumores neuroendocrinos, cuya denominación depende de su aparición en la glándula suprarrenal (feocromocitoma) o en los ganglios del sistema nervioso autónomo (paraganglioma). Hasta el 90% de los casos se dan en población adulta.
En el niño representan el 0,5% de los tumores del sistema nervioso periférico, y aproximadamente el 70% de los casos se asocian a diferentes síndromes genéticos, como la neoplasia endocrina múltiple tipo 2 (MEN2) –relacionada con la mutación del oncogén RET–, la enfermedad de Von Hippel Lindau (VHL) o la Neurofibromatosis tipo 1 (NF1). Las formas familiares se asocian a mutaciones de subunidades de la succinato dehidrogenasa (SDH).
Sus manifestaciones son catecolaminérgicas, con: hipertensión, taquicardia, sudoración, cefalea, palpitaciones, ansiedad/nerviosismo o temblores.
El diagnóstico es radiológico, mediante TC/RMN. Es obligada la realización de una MIBG, ya que pueden ser captantes y esto tiene relevancia terapéutica. Otros trazadores utilizados son: la 18F-fluorodopa, la 18F-fluorodopamina, los derivados del ácido tetracarboxílico (DOTA) marcados con 68Ga o el 111In-pentetreótido (Octreoscan).
Los metabolitos de catecolaminas en orina suelen estar elevados. También, puede detectarse el metabolito de la dopamina metoxitiramina, marcador de malignidad, en plasma.
El tratamiento tanto del feocromocitoma como del paraganglioma es quirúrgico, previo bloqueo adrenérgico (primero alfa y después beta, para evitar crisis hipertensivas).
En caso de presentación metastásica, en tumores captantes de MIBG, se puede valorar radioterapia dirigida, con 131I. La misma estrategia se está desarrollando con otros isótopos, como el 177Lu-, 90Y- o 111In-DOTA. En casos no captantes, las alternativas terapéuticas son menos efectivas. Se puede intentar radiofrecuencia, radioterapia y diversos regímenes de quimioterapia, para intentar controlar la enfermedad. Existen descritos casos que han permanecido estables durante largos periodos sin tratamiento. La terapia dirigida con Sunitinib, inhibidor de tirosín kinasa dirigido contra el receptor del factor de crecimiento del endotelio vascular, ha conseguido respuesta en algunos trabajos(20).
Función del pediatra de Atención Primaria
El neuroblastoma es preferentemente un tumor abdominal y del niño pequeño. Se debe sospechar ante toda palpación de una masa abdominal; sin embargo, este suele ser un hallazgo tardío.
Es importante incluir el neuroblastoma en el diagnóstico diferencial de cuadros aparentemente relativos a otras especialidades, como: hipertensión arterial inexplicada o déficits neurológicos, especialmente paraparesia o pérdida del control de esfínteres de aparición aguda.
A menudo, se refieren a Neurología a los pacientes con síntomas sugestivos de síndrome de opsoclono-mioclono (OMS): temblores, retraso psicomotor, movimientos oculares. Aunque infrecuente (2%), la asociación entre el OMS y el neuroblastoma es típica. Otros síntomas típicos son: el síndrome de Bernard-Claude-Horner en neuroblastomas cervicales, la aparición de varicocele/linfedema en extremidades inferiores y los hematomas orbitarios (en ojo de mapache).
La realización de dos pruebas sencillas y asequibles, como son: radiografía de tórax y ecografías cervical y abdominal, ayudará enormemente a orientar el caso. Los casos con alto índice de sospecha, deben remitirse de inmediato a centros especializados con Unidades de Oncología Pediátrica.
Bibliografía
Los asteriscos reflejan el interés del artículo a juicio del autor.
1. U.S. Cancer Statistics Working Group. United States Cancer Statistics: 1999-2012 Incidence and Mortality Web-based Report. Atlanta: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute; 2015. Disponible en: www.cdc.gov/uscs.
2. Peris-Bonet R, Salmerón D, Martínez-Beneito MA, et al. Childhood cancer incidence and survival in Spain. Ann Oncol. 2010; 21: 103-10.
3. Gatta G, Botta L, Rossi S, et al. Childhood cancer survival in Europe 1999-2007: results of EUROCARE-5 -a population-based study. Lancet Oncology. 2014; 15: 35-47.
4. Jiang M, Stanke J, Lahti JM. The connections between neural crest development and neuroblastoma. Curr Top Dev Biol. 2011; 94: 77-127.
5.*** Maris JM, Hogarty MD, Bagatell R, et al. Neuroblastoma. Lancet. 2007; 369: 2106-20.
6.** Hero B, Schleiermacher G. Update on Pediatric Opsoclonus Myoclonus Syndrome. Neuropediatrics. 2013; 44: 324-9.
7. Matthay KK, Shulkin B, Ladenstein R. Criteria for evaluation of disease extent by (123)I-metaiodobenzylguanidine scans in neuroblastoma: a report for the International Neuroblastoma Risk Group (INRG) Task Force. Br J Cancer. 2010; 102: 1319-26.
8. Beiske K, Burchill SE, Cheunget IY, et al. Consensus criteria for sensitive detection of minimal neuroblastoma cells in bone marrow, blood and stem cell preparations by immunocytology and QRT-PCR: recommendations by the International Neuroblastoma Risk Group Task Force. Br J Cancer. 2009 ; 100: 1627-37.
9. Sawada T, Matsumura T, Kawakatsu H, et al. Long-term effects of mass screening for neuroblastoma in infancy. Am J Pediatr Hematol Oncol. 1991; 13: 3-7.
10. Shimada H, Ambros IM, Dehner LP, Hata J, Joshi VV, Roald B. Terminology and morphologic criteria of neuroblastic tumors: recommendations by the International Neuroblastoma Pathology Committee. Cancer. 1999; 86: 349-63.
11.** Bagatell R, Cohn SL. Genetic discoveries and treatment advances in neuroblastoma. Curr Opin Pediatr. 2016; 28: 19-25.
12. Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer. 2003; 3: 203-16.
13. Carén H, Kryh H, Nethander, et al. High-risk neuroblastoma tumors with 11q-deletion display a poor prognostic, chromosome instability phenotype with later onset. PNAS. 2010; 107: 4323-8.
14. Carpenter EL, Mossé YP. Targeting ALK in neuroblastoma –preclinical and clinical advancements. Nat. Rev. Clin. Oncol. 2012; 9: 391-9.
15.*** Cohn SL, Pearson AD, London WB, et al. The International Neuroblastoma Risk Group (INRG) Classification System: An INRG Task Force Report. J Clin Oncol. 2008; 27: 289-97.
16. London WB, Boni L, Simon T, et al. The role of age in neuroblastoma risk stratification: the German, Italian, and children’s oncology group perspectives. Cancer Lett. 2005; 228: 257-66.
17.*** Yu AL, Gilman AL, Fevzi Ozkaynak F, et al. Anti-GD2 Antibody with GM-CSF, Interleukin-2, and Isotretinoin for Neuroblastoma.. N Engl J Med. 2010; 363: 1324-34.
18. Matthay KK, George RE, Yu AL. Promising therapeutic targets in neuroblastoma. Clin Cancer Res. 2012; 18: 2740-53.
19. Pinto NR, Applebaum MA, Volchenboum SL, et al. Advances in Risk Classification and Treatment Strategies for Neurorblastoma. J Clin Onc. 2015; 33: 3008-17.
20.** Martucci VL, Pacak K. Pheochromocytoma and paraganglioma: Diagnosis, genetics, management, and treatment. Curr Probl Cancer. 2014; 38: 7-41.
Bibliografía recomendada
- Maris JM, Hogarty MD, Bagatell R, et al. Neuroblastoma. Lancet. 2007; 369: 2106-20.
Una revisión breve, concisa y completa del neuroblastoma, que continúa estando vigente a pesar de haber pasado casi 10 años desde su publicación.
- Hero B, Schleiermacher G. Update on Pediatric Opsoclonus Myoclonus Syndrome. Neuropediatrics. 2013; 44: 324-9.
Actualización sobre el síndrome de opsoclono-mioclono, que incluye recomendaciones para su diagnóstico y tratamiento.
- Bagatell R, Cohn SL. Genetic discoveries and treatment advances in neuroblastoma. Curr Opin Pediatr. 2016; 28: 19-25.
Muy reciente actualización sobre el estado actual y las directrices futuras en el manejo del neuroblastoma.
- Cohn SL, Pearson AD, London WB, et al. The International Neuroblastoma Risk Group (INRG) Classification System: An INRG Task Force Report. J Clin Oncol. 2008; 27: 289-97.
Revisión de más de 8.000 casos de neuroblastoma con un detallado análisis de los factores pronósticos, que llevó a cambiar el sistema de estadificación del neuroblastoma.
- Yu AL, Gilman AL, Fevzi Ozkaynak F, et al. Anti-GD2 Antibody with GM-CSF, Interleukin-2, and Isotretinoin for Neuroblastoma.. N Engl J Med. 2010; 363: 1324-34.
Trabajo que impulsó la incorporación de la inmunoterapia al tratamiento de primera línea. Supervivencia significativamente superior (66 vs 46%) en los pacientes tratados con anticuerpo antiGD2 e inmunoestimuladores (IL2, GM-CSF).
- Martucci VL, Pacak K, Pheochromocytoma and paraganglioma: Diagnosis, genetics, management, and treatment. Curr Probl Cancer. 2014; 38: 7-41.
Revisión exhaustiva actualizada de ambas patologías. Establece criterios definidos para el diagnóstico genético (SDH, VHL, MEN2B, NF1). Revisa el tratamiento disponible, incluidas terapias dirigidas contra tirosín-kinasas (suritinib).
| Caso clínico |
|
Anamnesis Paciente de 16 meses de edad que consulta por temblores de dos semanas de evolución, mirada “extraña” y desde hace 3 días incapacidad para la marcha. Es una niña previamente sana, con desarrollo psicomotor normal hasta ese momento (bipedestación a los 10 meses y marcha autónoma a los 12), hija única de padres sanos no consanguíneos. Exploración física Buen estado general. Irritable, llanto continuo. Bien hidratada, nutrida y perfundida. No aspecto séptico ni distrófico. Fija la mirada y sigue objetos, pero tiene nistagmo horizontal. Auscultación pulmonar normal. Abdomen normal, con exploración dificultosa por llanto, pero sin peritonismo ni megalias. Sedestación normal, pero se niega a caminar. Al ofrecerle un bolígrafo, se observa temblor intencional. Constantes (FC, TA, SatO2) normales. Pruebas complementarias Hemograma, bioquímica e ionograma normales, exceptuando LDH de 850 U/l. Perfil férrico: ferritina de 1.500 mg/dl, resto normal. TC craneal: sin hallazgos. Eco abdominal: normal. Rx tórax: impresiona de condensación retrocardiaca. Se complementa el estudio con enolasa neuroespecífica: 145 mg/dl (normal <16 mg/dl) y RMN cérvivo-tóraco-abdominal que objetiva una masa de 4 x 6 cm dorsal paravertebral izquierda, que engloba sin comprimir grandes vasos. Se solicita MIBG con captación de la masa torácica descrita, sin otras captaciones.
|

















































































 y células ganglionares maduras con abundante citoplasma (flecha recta) en un GN")



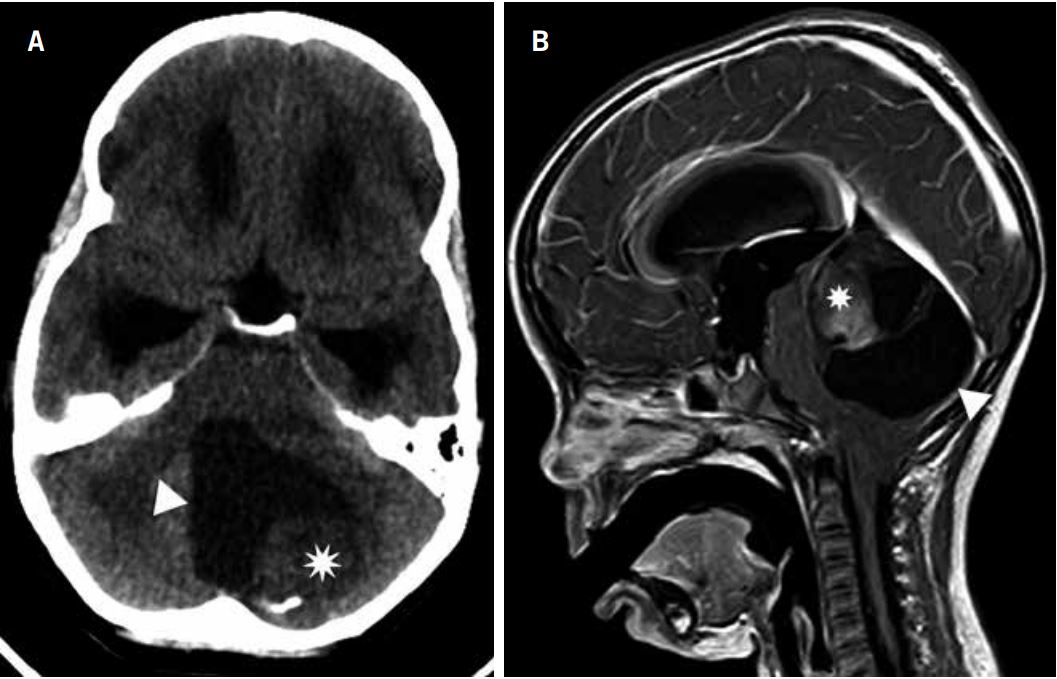
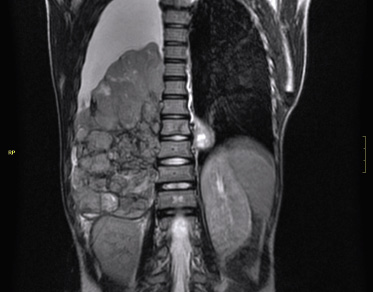
 RMN sagital del abdomen mostrando una masa anterior a la columna lumbar. B) Corte axial en T1 que muestra una masa que crece entre el músculo psoas dcho. y la columna.C) Corte axial en T2 del mismo nivel, mostrando una masa con señal de alta intensidad.")




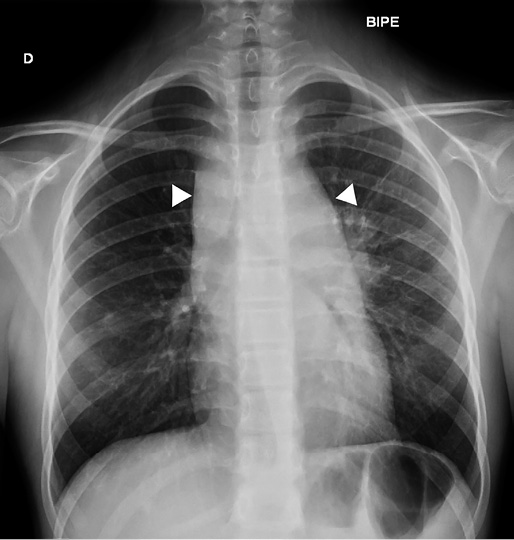
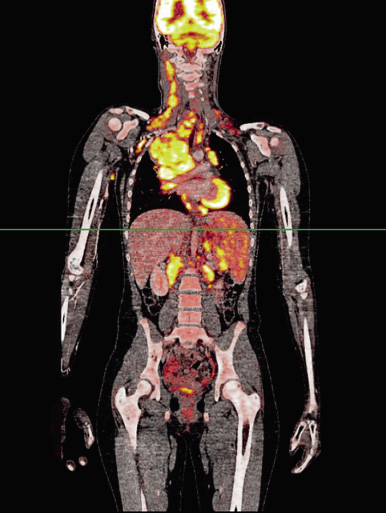
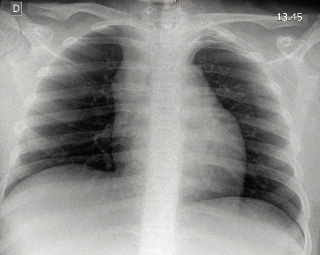
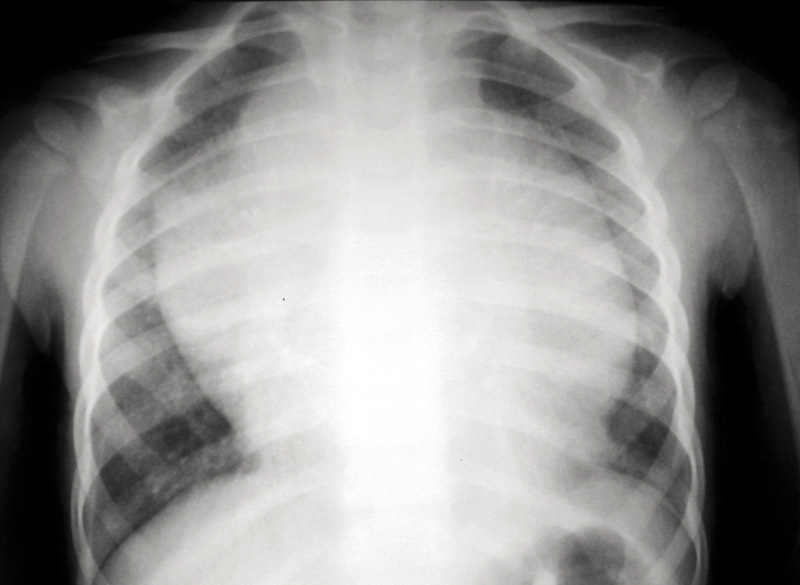
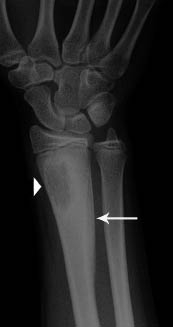
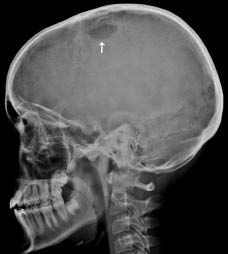
 Rx de tórax: imagen de condensación retrocardiaca en forma de huso. B) Gammagrafía con MIBG: depósito patológico de trazador en región retrocardiaca a nivel de la tumoración existente en esa zona. Además, en calota craneal, vértebras dorsales, pala ilíaca izquierda, húmeros, fémures y 6º arco costal en hemitórax")
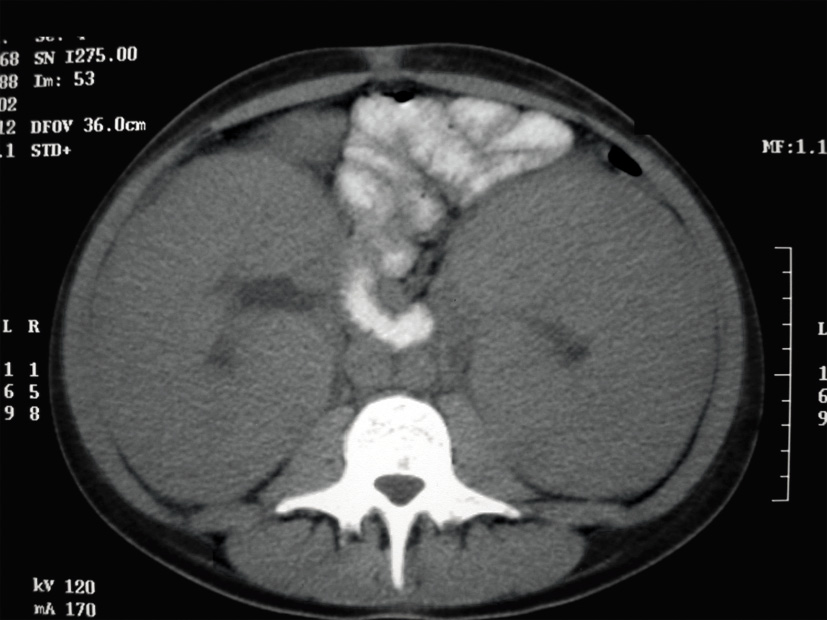
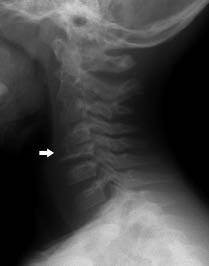
 toracoabdominal y retrocrural con presencia de calcificaciones en su interior. Afectación ósea en D7-D8-D9-D10 y L5 sin invasión del canal medular.")
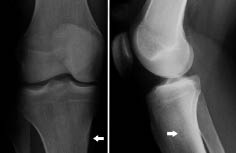
 Cilindro óseo que presenta infiltración neoplásica en forma de pequeños islotes celulares a nivel inter y peritrabecular en algunas celdillas. Las células tumorales presentan núcleos ovoides hipercromáticos y citoplasmas pobremente definidos. B) Con técnicas de inmunohistoquímica fueron enolasa +.")
































































