 |
| Temas de FC |
F. Villarejo Ortega, A. Aransay García, T. Márquez Pérez
Servicio de Neurocirugía del Hospital Infantil Universitario Niño Jesús, Madrid
| Resumen
Los tumores del sistema nervioso central en los niños representan la segunda causa más frecuente de tumores malignos. Actualmente, suponen una importante causa de morbi-mortalidad a pesar de los avances tecnológicos en: neuroimagen, neurocirugía, radioterapia y quimioterapia. La anamnesis y exploración clínica en Atención Primaria es fundamental para detectar precozmente los síntomas y signos de un tumor cerebral, muchas veces inespecíficos en esta edad, aunque la confirmación diagnóstica requiere la realización de un TC y/o una RM craneal. La cirugía es la base del tratamiento de los tumores cerebrales infantiles, puesto que permite establecer un diagnóstico anatomopatológico, en algunos casos, lograr una resección completa y, en otros casos, irresecables, reducir el volumen tumoral para aumentar la eficacia posterior de la quimioterapia y radioterapia. Frecuentemente, en el tratamiento de estos tumores, se requiere un abordaje multidisciplinar interviniendo: neurocirujanos, oncólogos, radioterapeutas, médicos rehabilitadores, neuropsicólogos y fisioterapeutas. En este artículo, se resumen algunos de los protocolos internacionales más recientes que han supuesto un avance en el tratamiento de los tumores cerebrales pediátricos y una mejoría de la supervivencia. Las investigaciones se están centrando en identificar marcadores genéticos que pueden condicionar el pronóstico y tratamiento de la enfermedad. |
| Abstract
Tumors of the central nervous system in |
Palabras clave: Tumores cerebrales pediátricos; Clasificación; Diagnóstico; Tratamiento
Key words: Pediatric brain tumors; Classification; Diagnosis; Treatment
Pediatr Integral 2016; XX (6): 401-411
Tumores cerebrales en niños
Introducción
Los tumores del sistema nervioso central (SNC) en los niños representan la segunda causa más frecuente de tumores malignos, por detrás de la leucemia, y se consideran los tumores sólidos más frecuentes en la edad pediátrica, suponiendo un 40-50% de todos los tumores.
En España, según el Registro Nacional de Tumores, cada año se registran 1.500 casos nuevos de cáncer infantil, de los cuales, un 20% corresponden a tumores del SNC.
A pesar de la mejora significativa en la tasa de curación de los tumores cerebrales pediátricos, durante las dos últimas décadas del siglo XX, como resultado de los avances tecnológicos en: neuroimagen, neurocirugía, radioterapia y quimioterapia, todavía suponen una importante causa de morbi-mortalidad. La falta de avances en el tratamiento de los tumores cerebrales pediátricos se debe fundamentalmente a la carencia de conocimientos sobre su patogénesis molecular de los tumores. Este déficit se está comenzando a superar gracias a las nuevas tecnologías que facilitan el entendimiento del paisaje genómico de los tumores cerebrales pediátricos, así como a la cooperación internacional entre laboratorios e investigadores clínicos(1).
Epidemiología
La incidencia anual de tumores del SNC en niños es de 2-5 casos por cada 100.000. Son ligeramente más frecuentes en varones(2), con una ratio varón/mujer de 1,5(3).
También, se han registrado pequeñas variaciones en la incidencia según la edad, siendo la incidencia mayor en los niños menores de 1 año(4). Se ha registrado una mayor incidencia en la raza blanca(3,4).
En los niños, predominan los tumores infratentoriales (55%), sobre los supratentoriales (45%), excepto en los 6 primeros meses de vida, que son más frecuentes los supratentoriales. Dentro de los tumores infratentoriales, los más frecuentes son: astrocitomas cerebelosos, gliomas troncoencefálicos y meduloblastomas, seguidos de los ependimomas. A nivel supratentorial, predominan los astrocitomas(5).
Los grupos histológicos más favorables son más frecuentes en adolescentes, registrándose una mayor supervivencia en los pacientes de entre 10 y 15 años, mientras que en los menores de 2 años, predominan los tumores de alto grado, con una localización más desfavorable, por lo que presentan un peor pronóstico y una menor supervivencia(3).
La supervivencia global a 5 años ha mejorado considerablemente en los últimos años, gracias al diagnóstico más precoz y a los avances en los tratamientos (cirugía, quimioterapia y radioterapia). Actualmente, la tasa de supervivencia varía entre el 35% y el 65% en función de diferentes factores, como son, fundamentalmente: la histología del tumor y su localización(3).
Etiología
Aunque la etiología de la mayoría de los tumores del SNC es desconocida, se han identificado diferentes factores de riesgo que a continuación detallamos, considerándose los más importantes los factores genéticos y la exposición a radiaciones ionizantes.
Factores genéticos
Síndromes de cáncer familiar
Existen diversos síndromes de cáncer familiar que aumentan la susceptibilidad a padecer tumores cerebrales. Menos del 10% de los niños con un tumor cerebral tienen un síndrome hereditario que les coloca en una situación de riesgo aumentado para el desarrollo de este tipo de tumores. Estos síndromes se caracterizan por la alteración en oncogenes y genes supresores de tumores, que desencadenan la aparición de tumores. Algunos de estos síndromes son(6): neurofibromatosis tipo 1, neurofibromatosis tipo 2, esclerosis tuberosa, síndrome de Von Hippel-Lindau, síndrome de Cowden, síndrome de Gorlin o del nevus basocelular, síndrome de Turcot, síndrome de Li-Fraumeni y retinoblastoma hereditario. La mayoría de estos síndromes tienen un patrón de herencia autosómica dominante y se han demostrado mutaciones genéticas específicas para cada una de ellas (Tabla I).

Los niños afectados por estas enfermedades hereditarias deben someterse a seguimientos periódicos para detectar precozmente el desarrollo de una neoplasia.
Historia familiar
Los hallazgos sobre el riesgo de tumores cerebrales en la infancia entre miembros de una familia varía sustancialmente entre los diferentes estudios(6). La presentación familiar de tumores el SNC es infrecuente, aparte de las neoplasias relacionadas con los síndromes neurocutáneos, pero se han descrito casos familiares de meningiomas y gliomas.
Radiaciones ionizantes
La exposición a radiaciones ionizantes intraútero, o directamente al utilizar la radioterapia craneal, es una causa bien documentada de tumores cerebrales en niños(6). En los niños con tinea capitis que fueron tratados con radioterapia durante los años 40 y 50, se ha visto un incremento de riesgo de desarrollo de gliomas y meningiomas 22 a 34 años después. Se han publicado diversos estudios que describen casos de tumores cerebrales en niños con leucemias linfoblásticas que, durante el tratamiento, recibieron radioterapia craneal, mostrando un claro aumento del riesgo de tumores cerebrales (gliomas y tumores neuroectodérmicos primitivos) en niños que recibieron irradiación profiláctica del SNC (25 Gy)(6). En estos casos, la latencia entre la radioterapia y el desarrollo del tumor cerebral se estima que fue de entre 7-9 años, con un mayor riesgo en niños de menor edad.
Inmunosupresión
En algunos síndromes de inmunodeficiencia, como el Wiskott-Aldrich, la ataxia-telangiectasia y la inmunodeficiencia adquirida, tras el trasplante de órganos sólidos pueden desarrollarse linfomas cerebrales con mayor frecuencia que en la población general(6).
Exposiciones ambientales
La influencia de diversas exposiciones ambientales en el desarrollo de los tumores cerebrales infantiles, ha sido estudiada por diversos investigadores. Los resultados de todos estos estudios no han sido concluyentes. Actualmente, no existe evidencia de aumento de riesgo de padecer tumores del SNC para ninguno de los factores ambientales estudiados, incluyendo: el uso de teléfonos móviles, la exposición a líneas de alta tensión, los traumatismos craneales, las nitrosaminas o nitrosoureas en tetinas de chupetes o biberones y otros factores de la dieta.
Clasificación
La clasificación de los tumores cerebrales se realiza en función de la estirpe celular a partir de la cual se origina el tejido anormal (Tabla II).

Además, cada tipo tumoral se subclasifica en diferentes grados (Tabla III) según las características histológicas (necrosis, neovascularización) y citológicas (mitosis, diferenciación celular).

Se prevé que la nueva clasificación de la Organización Mundial de la Salud (OMS) incluya marcadores genéticos, dada la importancia que se ha visto que tienen en el pronóstico de la enfermedad (Fig. 1)(7).

Figura 1. Clasificación de los gliomas infiltrantes según las alteraciones moleculares IDH: gen que codifica la isocitrato deshidrogenasa.
Conocer el tipo de tumor es fundamental, porque permite no solo predecir la historia natural de la enfermedad, sino también, la respuesta al tratamiento y el pronóstico.
Los astrocitomas, tumores derivados de las células gliales tipo astrocito, representan los tumores del SNC más frecuentes en la edad pediátrica, suponiendo hasta el 50% de los casos. Existen distintos tipos de astrocitomas según sus características histológicas, citológicas y agresividad. Los astrocitomas pilocíticos (Grado I de la OMS) son los más frecuentes y representan hasta el 25% de los tumores del SNC pediátricos(8). Estos tumores se consideran benignos y son más frecuentes en la fosa posterior, aunque pueden aparecer en cualquier localización (Fig. 2).
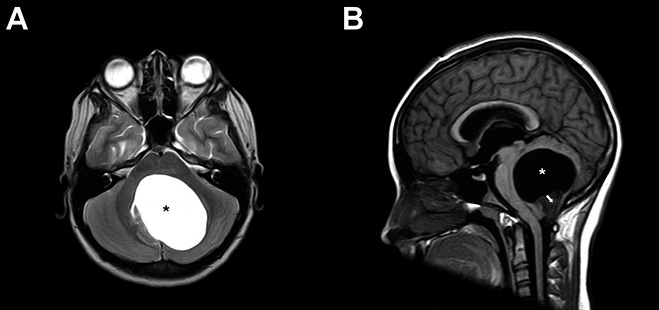
Figura 2. Astrocitoma pilocítico en RM craneal. Tumoración en hemisferio cerebeloso izquierdo con un componente quístico (*) y un componente sólido en su porción más caudal (→), que colapsa el acueducto de Silvio y el IV ventrículo. A. Corte axial en T2. B. Corte sagital en T1.
Otras localizaciones frecuentes de los gliomas en niños son: el hipotálamo, las vías ópticas y el tronco del encéfalo. Típicamente, estas lesiones suelen ser de bajo grado (Grado I y II de la OMS), pero su localización hace que sea difícil realizar una extirpaciónquirúrgica completa y, por lo tanto, la morbilidad es alta y el pronóstico malo, aunque se trate de neoplasias de bajo grado.
El segundo tumor del SNC, más frecuente en niños es el meduloblastoma (Fig. 3).

Figura 3. Meduloblastoma en RM craneal con contraste. Tumoración en IV ventrículo (*) con realce intenso y heterogéneo y una captación leptomeníngea difusa de predominio en fosa posterior izquierda compatible con diseminación leptomeníngea (→). A. Corte sagital en T1. B. Corte axial en T1.
Este tumor se origina en el IV ventrículo, a partir de precursores de células madre neurales, y suponen entre el 15-20% de los tumores cerebrales pediátricos(3).
Otros tumores frecuentes en edad pediátrica, derivados de células neuroepiteliales, son: ependimomas (10% de los tumores pediátricos del SNC), gangliogliomas (2,5%) y tumores neuroectodérmicos primitivos (1,9%).
En cuanto a las neoplasias pediátricas no neuroepiteliales, las más frecuentes son: los craneofaringiomas (5,6%), los tumores de células germinales (2,5%), localizados en la región pineal o en otras localizaciones y pueden ser: germinomas, teratomas, coriocarcinomas y tumores del seno endodérmico, tumores teratoides/rabdoides atípicos (1,3%) y tumores de plexos coroideos (0,9%).
La incidencia de los distintos tipos de tumores del SNC en niños, varía según el grupo de edad(3). Así, entre los 0 y los 2 años, predominan: papilomas de plexos coroideos, astrocitomas desmoplásicos, teratomas, tumores neuroectodérmicos primitivos (PNETs) y tumores teratoides/rabdoides atípicos. Entre los 3 y los 11 años, los astrocitomas y craneofaringiomas son los más frecuentes. A partir de los 12 años, son frecuentes los tumores de células germinales, mientras que los craneofaringiomas son raros(3).
La distribución de los distintos tipos de astrocitomas también varía con la edad. Los astrocitomas pilocíticos predominan, sobre todo, en el grupo de 3 a 11 años y en la adolescencia temprana, pero, a partir de los 15 años, la incidencia de los astrocitomas pilocíticos cae y son más frecuentes los astrocitomas grado III y IV de la OMS.
Clínica
El diagnóstico de tumor cerebral puede ser muy difícil en los niños. Frecuentemente, los niños, sobre todo los más pequeños, son incapaces de referir sus síntomas. Por otra parte, el curso puede ser insidioso y con manifestaciones poco específicas, tales como: cambios de carácter, irritabilidad o vómitos.
Incluso para los padres, ciertos cambios pueden pasar desapercibidos. Los síntomas y signos de los tumores intracraneales en el niño dependen de la edad, de la localización del tumor y de la presencia o ausencia de hipertensión intracraneal.
Hipertensión intracraneal
La cefalea es un síntoma prácticamente constante en los tumores cerebrales infantiles. Puede ir acompañada de vómitos, que suelen ser matutinos o nocturnos, e ir o no precedidos de náuseas.
En el lactante, puede manifestarse como irritabilidad o llanto inconsolable. En pacientes pediátricos con cefalea, los siguientes datos tienen valor predictivo como indicadores de proceso expansivo intracraneal: cefalea durante el sueño, historia corta (<6 meses) de dolor de cabeza, asociación con confusión o vómitos, síntomas visuales y presencia de signos neurológicos anormales. Gran parte de los niños con hipertensión intracraneal presentan papiledema, pero su ausencia no excluye la presencia de hipertensión intracraneal. En recién nacidos y lactantes, hay dos datos muy fiables de hipertensión intracraneal: la macrocefalia y la tensión de la fontanela. En estas edades, la parálisis del VI par craneal aislada, es un síntoma de hipertensión intracraneal y no de focalidad.
En un alto porcentaje de tumores cerebrales infantiles, la hipertensión intracraneal se debe a hidrocefalia, siendo más frecuente en los tumores de la región pineal-lámina cuadrigémina, seguidos de los infratentoriales, los de la región hipotalámica y, finalmente, de los hemisféricos.
Síntomas focales
Los síntomas y signos de focalidad son más evidentes cuanto mayor es la edad del niño. Este hecho se debe a la inmadurez del cerebro de los niños muy pequeños, que se refleja en una falta de expresividad clínica. Estos síntomas dependen de la localización del tumor. A veces, en lactantes, una pérdida de visión importante puede pasar totalmente desapercibida hasta que la lesión está muy avanzada.
La hemiparesia, hipertonía e hiperreflexia son las manifestaciones más frecuentes en los tumores supratentoriales y, en menor proporción, los trastornos de la sensibilidad. En los infratentoriales, los síntomas y signos más habituales son: diplopía, ataxia y nistagmus.
La primera manifestación de un tumor de fosa posterior puede ser una tortícolis. En los tumores de tronco, puede haber parálisis de pares craneales y afectación de vías largas.
Epilepsia
Las crisis epilépticas pueden constituir la primera, o única, manifestación de un tumor cerebral.
Las crisis pueden ser generalizadas, focales o psicomotoras, y pueden ir seguidas de una parálisis residual postcrítica. Un trazado electroencefalográfico de afectación focal debe sugerir la realización de otros estudios complementarios para descartar un origen orgánico de las crisis, aunque hay tumores cerebrales que no producen alteraciones. Los tumores infratentoriales pueden producir enlentecimiento bilateral de la actividad cerebral.
Síntomas inespecíficos
Los cambios de personalidad, las alteraciones emocionales y la disminución del rendimiento escolar pueden ser manifestaciones de un tumor cerebral. En niños muy pequeños, la detención o el retroceso del desarrollo psicomotor sugieren una lesión orgánica y requieren un estudio más amplio.
Diagnóstico
La anamnesis y exploración clínica son fundamentales para detectar síntomas y signos de alarma que nos hagan sospechar la posible presencia de un tumor cerebral y, por lo tanto, solicitar las pruebas necesarias.
Pruebas de imagen
La tomografía computarizada (TC) y la resonancia magnética (RM) son la base del diagnóstico, tratamiento, planificación quirúrgica y seguimiento. La adición de contraste intravenoso a estos estudios permite detectar la alteración en la barrera hematoencefálica, que es típico en los tumores malignos del sistema nervioso central, así como también identificar lesiones sutiles y delimitar los bordes de la lesión. Hay que tener en cuenta que en los gliomas pediátricos, lesiones de bajo grado, como los astrocitomas pilocíticos, a menudo, captan contraste a pesar de ser lesiones benignas(3).
Tomografía computarizada (TC)
La TAC se puede obtener en el servicio de urgencias y proporciona, de forma rápida, datos sobre: la presencia o ausencia de tumor, el tamaño del mismo, la forma y densidad tumoral, la localización, el comportamiento tras la administración de contraste, presencia de calcificaciones, zonas de necrosis y quistes, edema peritumoral, desplazamientos y herniaciones cerebrales, afectación de estructuras óseas, presencia de hidrocefalia y hemorragia tumoral, etc.
Resonancia magnética (RM)
Las principales ventajas de la RM en el estudio de las neoplasias cerebrales son su capacidad multiplanar y la alta resolución de la imagen. El manejo de diferentes parámetros y secuencias nos permite un mejor estudio de las características de la lesión, así como diferenciar los distintos tejidos del SNC. Las posibles desventajas de la RM son el largo tiempo de exploración y la degradación de la imagen si existe movimiento. Por lo tanto, se exige una gran colaboración por parte del paciente, obligando frecuentemente a la sedación o anestesia, para garantizar la calidad del estudio sin artefactos por movimiento. Otra desventaja de la RM es que, al someterse el paciente a un campo magnético intenso, puede estar contraindicada en algunos pacientes que porten componentes ferromagnéticos o cuerpos extraños metálicos.
En los últimos años, diferentes avances tecnológicos (tanto mejoras en la configuración de hardware, como en el desarrollo de nuevas secuencias del pulso) permiten nuevas técnicas que mejoran la detección y caracterización de los tumores. Entre estos nuevos avances, destacamos las técnicas de RM que aportan información metabólica (RM difusión-perfusión y espectroscopia por RM) y funcional (RM funcional para valorar áreas de activación del córtex cerebral en zonas elocuentes).
Algunos tumores cerebrales, como los meduloblastomas, los tumores de células germinales, los ependimomas y los PNETs, son propensos a diseminarse dentro del SNC. Por lo tanto, si se sospecha algunos de estos tumores, se debe ampliar el estudio de neuroimagen al resto del neuroeje, para detectar precozmente metástasis (Figs. 3 y 4).

Figura 4. PNET en RM craneal (A) con metástasis espinales en RM de columna (B). Tumoración centrada en protuberancia y mesencéfalo (*). No presenta realce significativo tras la administración de contraste. Nódulo espinal con captación de contraste (→) a nivel L1-L2 compatible con metástasis. A. Corte sagital en T1 con contraste (craneal). B. Corte sagital en T1 con contraste (columna).
Se recomienda que la RM de columna se realice antes de la intervención quirúrgica, para disminuir el número de falsos positivos secundarios a restos de sangre. Si la RM de columna se realiza después de la cirugía, es recomendable esperar, al menos, dos semanas(3).
Angiografía cerebral
La realización de una angiografía cerebral está indicada en tumores con gran vascularización para conocer los vasos afluentes a la neoplasia, sobre todo, si se contempla la posibilidad de embolización preoperatoria como parte del tratamiento.
Punción lumbar
La punción lumbar se realiza después de las pruebas de imagen, en los tumores con tendencia a diseminar. El objetivo es realizar un examen citológico y detectar células tumorales en líquido cefalorraquídeo (LCR), imposibles de identificar en ninguna prueba de imagen. La presencia o ausencia de células tumorales en LCR, condiciona la asignación a una determinada categoría de tratamiento.
Marcadores tumorales
En determinados tumores de la región pineal y en algunos tumores de estirpe embrionaria, la determinación de marcadores tumorales en sangre y/o LCR es importante para la orientación sobre el tipo tumoral, valorar el grado de resección, la respuesta al tratamiento y la presencia de recidiva tumoral. Los marcadores que se determinan más frecuentemente son: alfa-fetoproteína, subunidad β de la gonadotropina coriónica y el antígeno cárcino-embrionario.
Valoración neuropsicológica
La valoración neuropsicológica es cada día más importante en la planificación de la cirugía tumoral, sobre todo en tumores de bajo grado que asientan sobre áreas cerebrales funcionalmente importantes, ya que pueden proporcionar información sobre los efectos adversos neuropsicológicos que está produciendo el tumor y la posibilidad de secuelas neuropsicológicas. Permiten también valorar el daño causado por la resección tumoral y los efectos adversos de los tratamientos coadyuvantes, fundamentalmente de la radioterapia y quimioterapia.
Tratamiento
Existen diversas opciones de tratamiento que, fundamentalmente, son: cirugía, quimioterapia y radioterapia, en sus diversas formas.
Hay que destacar que el tratamiento de los tumores del SNC en los niños requiere un abordaje en equipo, coordinando las diversas especialidades que colaboran en el tratamiento mediante comités de neuro-oncología pediátrica. En la tabla IV(9-16), se resumen los protocolos y estudios institucionales más recientes que han supuesto un avance en el tratamiento de los tumores cerebrales pediátricos y una mejoría de la supervivencia.


Cirugía
La cirugía cumple dos objetivos principales: 1) establecer un diagnóstico de certeza; y 2) reducir el volumen tumoral. La citorreducción parece esencial para la ulterior eficacia de la radioterapia y quimioterapia.
Abordaje directo del tumor
Para la mayoría de los tumores, la opción preferida es el abordaje directo, con intención de exéresis total (Fig. 5), o lo más amplia posible, dependiendo de la naturaleza del tumor y de su localización.

Figura 5. Papiloma de plexo coroideo en RM craneal. Masa en ventrículo lateral izquierdo (*) con intensa captación de contraste extirpada completamente en tres tiempos. A. Corte sagital en T1 con contraste pre operatorio. B. Corte sagital en T1 con contraste tras primera intervención. C. Corte sagital en T1 con contraste tras segunda intervención. D. Corte sagital en T1 con contraste tras tercera y última intervención.
Para conseguir estos fines, disponemos de una serie de técnicas coadyuvantes, como son: la microcirugía, técnicas de cirugía mínimamente invasiva, localización intraoperatoria tumoral mediante ultrasonidos, registros neurofisiológicos intraoperatorios, neuronavegación, láser, aspirador ultrasónico, TC o RM intraoperatorias, etc.
Biopsia estereotáxica o guiada por neuronavegación
En ciertos tumores de localización profunda (tronco del encéfalo, ganglios basales o tálamo), puede estar indicada la biopsia estereotáxica o guiada por neuronavegador, que puede ir o no seguida de cirugía más amplia.
Tratamiento quirúrgico de la hidrocefalia
Existen distintas opciones de tratamiento: a) colocación de drenaje ventricular externo temporalmente hasta que se extirpe la lesión tumoral que obstruye la circulación de LCR; b) inserción preoperatoria o postoperatoria de una derivación permanente de LCR (válvula ventrículo-peritoneal o ventrículo-atrial); y c) ventrículo cisternostomía del suelo del III ventrículo mediante neuroendoscopia. El empleo de la derivación permanente de LCR es objeto de debate, puesto que pueden presentar complicaciones como la diseminación tumoral por la derivación y la herniación transtentorial ascendente, en casos de tumores de fosa posterior.
Radioterapia
La radioterapia local está indicada en tumores malignos, en propensos a recidivar, en exéresis incompletas, e incluso en tumores de bajo grado irresecables.
La tendencia es radiar el lecho tumoral y el borde adyacente. Algunos autores discuten la conveniencia de administrar radioterapia al eje craneoespinal cuando no hay evidencia de diseminación por los riesgos que comporta, especialmente teniendo en cuenta la posibilidad de diagnosticar la diseminación mediante técnicas poco invasivas, como la citología del LCR y la RM con contraste. Van apareciendo diversas modalidades de radioterapia, como son la radioterapia hiperfraccionada o la radiocirugía estereotáxica, cada vez más precisas, que permiten proporcionar altas dosis de radiación, pero solo en el tejido tumoral, para disminuir los efectos secundarios derivados de la irradiación de zonas sanas. Debido a los daños irreparables que la radioterapia puede ocasionar en el SNC en desarrollo de los niños, la tendencia es a diferir, siempre que sea posible, su utilización hasta después de los 3 años de edad.
Quimioterapia
Tradicionalmente, la quimioterapia se empleaba solo en tumores malignos con alto riesgo, como el meduloblastoma y los astrocitomas de alto grado. En los últimos años, se está utilizando quimioterapia en gliomas de bajo grado incompletamente extirpados, o tras la progresión tumoral, con resultados prometedores en ciertos casos.
La quimioterapia puede seguir a la radioterapia o puede precederla. La ventaja de la quimioterapia postoperatoria es que se aprovecha la ruptura de la barrera hematoencefálica, producida por la cirugía. La quimioterapia es más eficaz antes de que la radioterapia produzca cambios vasculares que impidan el paso de estas drogas. Sin embargo, la toxicidad de los citostáticos es menor si se administra después de la radioterapia; por lo que, se toleran dosis más altas y repetidas.
Se utilizan estimulantes hemotopoyéticos para disminuir la mielotoxicidad de ciertos agentes. En niños menores de 3 años, la tendencia actual es diferir la radioterapia, administrando mientras tanto, medicamentos citostáticos. Un nuevo abordaje es el autotrasplante de médula ósea en combinación con diversos tratamientos quimioterápicos.
Otras técnicas, aún en estudio, son: la inmunoterapia y la terapia génica.
Bibliografía
Los asteriscos reflejan el interés del artículo a juicio del autor.
1.** Gajjar A, Bowers DC, Karajannis MA, et al. Pediatric Brain Tumors: Innovative Genomic Information Is Transforming the Diagnostic and Clinical Landscape. J Clin Oncol. 2015; 33: 2986-98.
2. Linabery AM, Ross JA. Trends in childhood cancer incidence in the U.S. (1992-2004). Cancer. 2008; 112: 416-32.
3.*** Reynolds R and Grant G. General approaches and considerations for pediatric brain tumor. En: Winn HR, ed. Youmans neurological surgery. Elsevier; 2011. p 2040-46.
4. Ostrom QT, de Blank PM, Kruchko C, et al. Alex’s Lemonade Stand Foundation Infant and Childhood Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2007-2011. Neuro Oncol. 2015; 16: x1-36.
5. Greenberg MS. Tumor. En: Greenberg MS, ed. Handbook of neurosurgery. New York. Thieme; 2010. p 582-768.
6.** Johnson KJ, Cullen J, Barnholtz-Sloan JS, et al. Childhood brain tumor epidemiology: a brain tumor epidemiology consortium review. Cancer Epidemiol Biomarkers Prev. 2014; 23: 2716-36.
7. Brat DJ, Verhaak RG, et al. Cancer Genome Atlas Research Network. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med. 2015; 372: 2481-98.
8. Villarejo F, Belinchon JM, Gomez de la Riva A. Prognosis of cerebellar astrocitomas in children. Childs Nerv Syst. 2008; 24: 203-10.
9. Packer RJ, Gajjar A, Vezina G, et al: Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. J Clin Oncol. 2006; 24: 4202-08.
10. Taylor RE, Bailey CC, Robinson K, et al: Results of a randomized study of preradiation chemotherapy versus radiotherapy alone for non metastatic medulloblastoma: The International Society of Paediatric Oncology/United Kingdom Children’s Cancer Study Group PNET-3 study. J Clin Oncol. 2003; 21:1581-91.
11. Tarbell NJ, Friedman H, Polkinghorn WR, et al: High-risk medulloblastoma: A Pediatric Oncology Group randomized trial of chemotherapy before or after radiation therapy (POG 9031). J Clin Oncol. 2013; 31: 2936-41.
12. Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005; 352: 987-96.
13. Cohen KJ, Pollack IF, Zhou T, et al: Temozolomide in the treatment of high-grade gliomas in children: A report from the Children’s Oncology Group. Neuro Oncol. 2011; 13: 317-23.
14. Cohen KJ, Heideman RL, Zhou T, et al. Temozolomide in the treatment of children with newly diagnosed diffuse intrinsic pontine gliomas: A report from the Children’s Oncology Group. NeuroOncol. 2011; 13: 410-16.
15. Bradley KA, Zhou T, McNall-Knapp RY, et al. Motexafin-gadolinium and involved field radiation therapy for intrinsic pontine glioma of childhood: A children’s oncology group phase 2 study. Int J Radiat Oncol Biol Phys. 2013; 85: e55-60.
16. Ater JL, Zhou T, Holmes E, et al: Randomized study of two chemotherapy regimens for treatment of low-grade glioma in young children: A report from the Children’s Oncology Group. J Clin Oncol. 2012; 30: 2641-47.
| Caso clínico |
|
Varón de 4 años que fue llevado al servicio de urgencias por presentar un cuadro de 8 meses de evolución de temblor en ambas manos al realizar movimientos finos, caídas frecuentes y dificultad para mover la pierna y el brazo derecho en el último mes. No había referido cefalea, ni náuseas o vómitos ni alteraciones del comportamiento. Antecedentes personales Embarazo controlado normal. Parto vaginal instrumentado (ventosa) a la 40+1 semanas, inducido por oligoamnios. Apgar: 9/10. No RAMc. Calendario vacunal correcto, incluido vacuna contra Neumococo y Rotavirus. Antecedentes familiares Sin interés para el proceso actual. Exploración neurológica GCS (Glasgow Coma Scale): 15. Pupilas isocóricas y normorreactivas. Pares craneales normales. Movimientos oculares extrínsecos conservados, sin nistagmus. Hemiparesia derecha (4/5), puede caminar sin ayuda. Dismetría en maniobra dedo-nariz. Romberg negativo. Fondo de ojo normal, sin papiledema ni otros hallazgos patológicos. Se le realizó un TC craneal urgente que evidenció un ensanchamiento difuso del tronco del encéfalo sospechoso de tumor, sin signos de hidrocefalia. Ingresó para iniciar tratamiento y completar estudio. Exploraciones complementarias • RM craneal con contraste intravenoso, angioRM y espectroscopía (Fig. 6). Tumoración sólido-quística bien definida, localizada en la vertiente izquierda del mesencéfalo y se extiende caudalmente hasta protuberancia superior izquierda. El polo sólido en el margen posterolateral de la tumoración presenta un realce heterogéneo tras la administración de contraste. No se observa restricción de la difusión. La lesión ejerce ligero efecto de masa con impronta y desplazamiento del III ventrículo. Discreto edema vasogénico perilesional. El estudio de espectroscopia RM del componente sólido de la lesión se observa un descenso del pico N-acetil-aspartato y discreta elevación de colina sin pico reseñable de lípidos o lactato. No se visualizan alteraciones en angio-RM de polígono de Willis. Sistema ventricular de tamaño normal y morfología conservada. • Potenciales evocados somatosensoriales: alteración de la conducción medular de los cordones posteriores tras estímulo del nervio tibial posterior derecho. Tratamiento Fue intervenido quirúrgicamente, realizándole craneotomía temporal izquierda, evacuación del quiste y biopsia de la porción sólida del tumor. Presentó buena evolución postquirúrgica, clínica y radiológica. Un mes y medio después de la cirugía sufrió empeoramiento de la paresia de pierna derecha, por lo que se realizó nueva RM craneal que objetivó un aumento de tamaño de la porción quística de la lesión mesencefálica. Se decidió reintervenir al paciente para evacuar el quiste e implantar un reservorio Ommaya para evitar nuevas reintervenciones. Se completó tratamiento con quimioterapia.
Figura 6. RM craneal en T1 con contraste. Tumoración con un componente quístico (*) y otro sólido (→) en su margen posterolateral con realce heterogéneo tras la administración de contraste. A. Corte sagital en T1. B. Corte coronal en T1.
|

