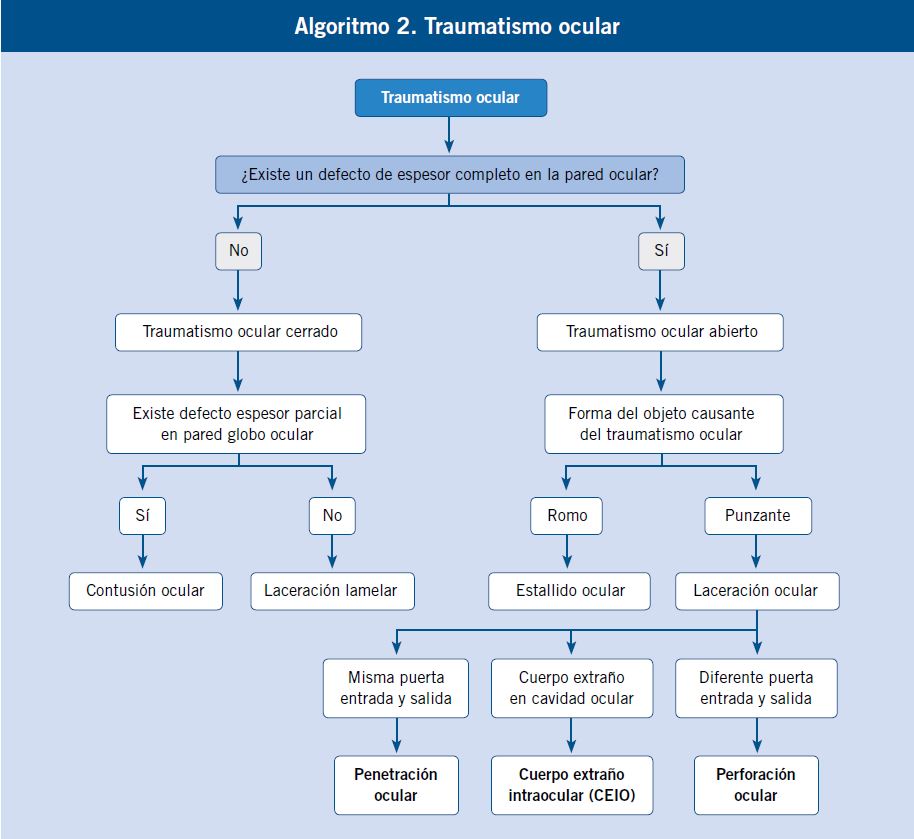
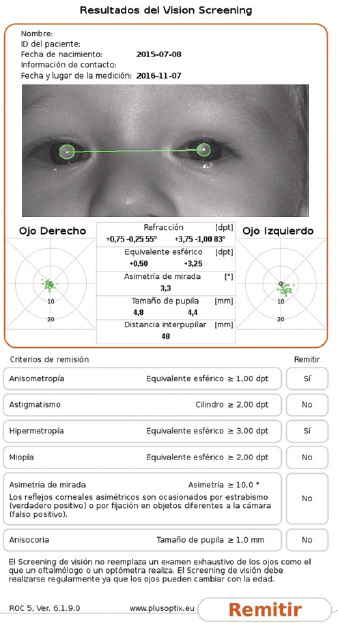
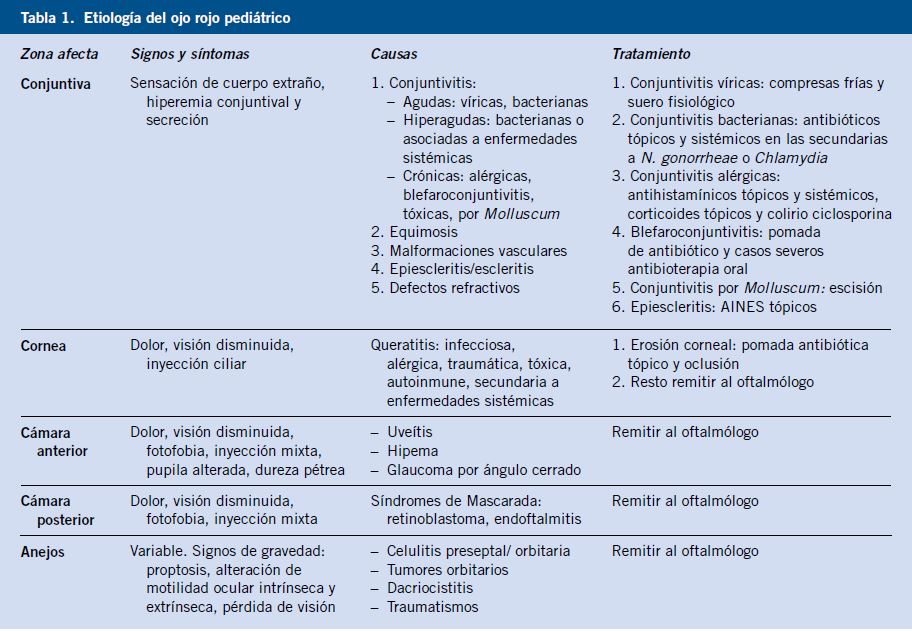
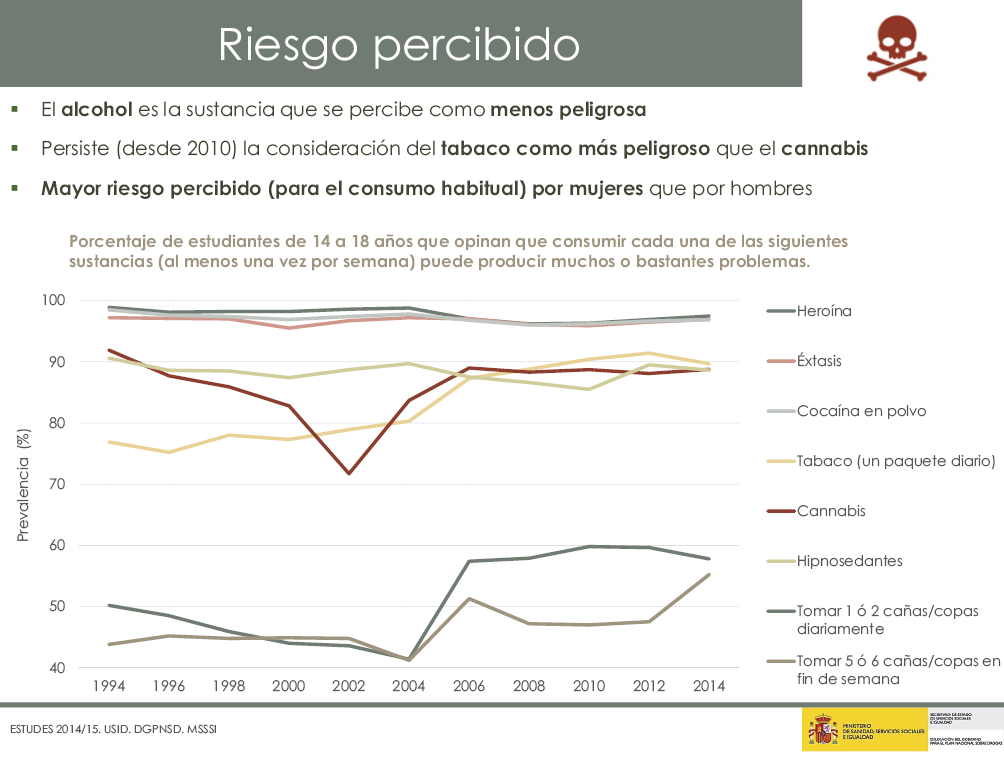
|
| Temas de FC |
V. Martín Gómez*, J.M. Casanovas Gordó**
*Oftalmóloga. Especialista en segmento anterior y córnea. Hospital Sant Joan Despi Moisès Broggi. Barcelona. **Pediatra. CAP Roquetes-Canteres. Institut Català de la Salut. Barcelona
| Resumen
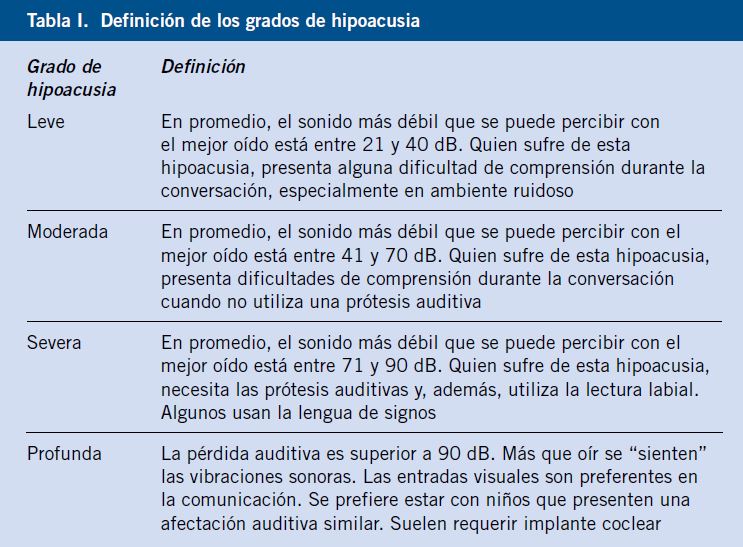
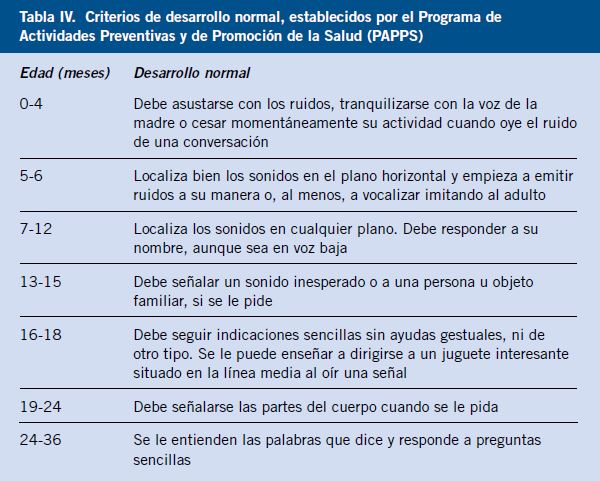
El pediatra de Atención Primaria es el primero en valorar los traumatismos oculares que se producen en el domicilio o en el colegio. Los niños preadolescentes y adolescentes suelen ser los más afectados. Se describe la información que corresponde a la historia clínica, la exploración del paciente, con los detalles que tienen mayor interés para el pediatra. La afectación del párpado, la conjuntiva, la córnea, el iris, el cristalino, el vítreo, la coroides y la retina, desde un punto de vista traumático. También se explican los datos más importantes que corresponden a la fractura de la órbita, la lesión del nervio óptico y la perforación ocular, que pueden dar lugar a graves secuelas. Los traumatismos oculares en el deporte y la afectación ocular en el niño maltratado, también son motivo de revisión. |
| Abstract
The primary care pediatrician is the first to assess eye injuries that occur at home or at school. Preteens and adolescents are often the most affected. It describes the information that corresponds to the Medical pacient history, the exploration of the patient with the details that are of greatest interest to the pediatrician. The involvement of the eyelid, conjunctiva, cornea, iris, lens, vitreous, choroid and retina, from a traumatic point of view. The most important data that correspond to the fracture of the orbit, the optic nerve injury and the ocular perforation that can lead to serious sequelae are also described. Ocular trauma in the sport and ocular involvement in the abused child are also a reason for revision. |
Palabras clave: Traumatismos oculares; Pediatría; Lesiones conjuntivales y corneales; Fractura de la órbita; Afectación ocular del niño maltratado
Key words: Ocular traumatism; Pediatrics; Conjunctival and corneal injuries; Fracture of the orbit; Ocular impairment of abused child
Pediatr Integral 2018; XXII (1): 45 – 57
Traumatismos oculares
Introducción
“Los traumatismos oculares deben ser considerados como una enfermedad”. JT Banta.(1) La primera causa de visita oftalmológica pediátrica de urgencia son las conjuntivitis y, la segunda, los traumatismos oculares.
El estrabismo es la causa más frecuente de intervención quirúrgica ocular en el niño y la ambliopía la de pérdida de visión monocular precoz. Después de ambas patologías, están los traumas oculares como causa más importante de morbilidad ocular infantil.
Los traumatismos oculares se distribuyen según las edades, con una primera incidencia en los últimos años de la adolescencia y una segunda en los pacientes mayores de 70 años de edad(1,2).
Las lesiones oculares graves son más frecuentes en los varones, con una proporción de 3-5/1. Son causa importante de pérdida de visión en los países menos desarrollados y en los estratos sociales menos favorecidos.
Son los responsables de ceguera unilateral, baja visión bilateral y ceguera bilateral. Uno de cada tres casos de ceguera en Pediatría es por traumatismo. El traumatismo ocular es además una enfermedad recurrente, tras un primer traumatismo existen 3 veces más probabilidades de sufrir un 2º trauma ocular.
Buena parte de los traumatismos son evitables. Es indudable que la supervisión de los adultos reduce la incidencia y el bajo nivel educativo o socioeconómico aumenta el riesgo de lesión ocular. El pediatra, muchas veces, será el primero en atenderlos.
En los primeros años de vida, la incidencia de traumatismos oculares es prácticamente igual en ambos sexos, pero esta tendencia cambia con una mayor frecuencia en varones en la población adulta. La mayoría de las lesiones se producen en el hogar o colegio(2).
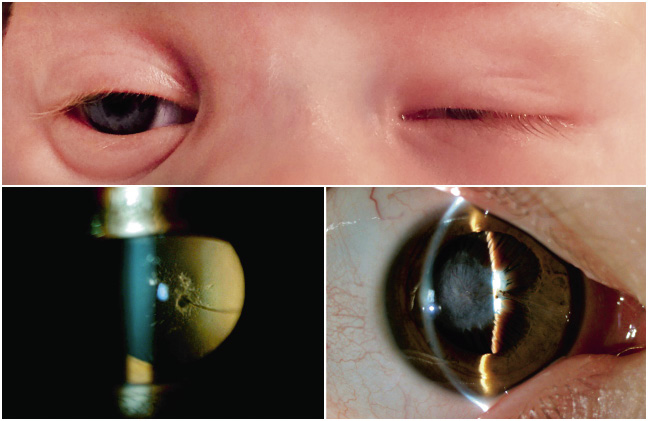
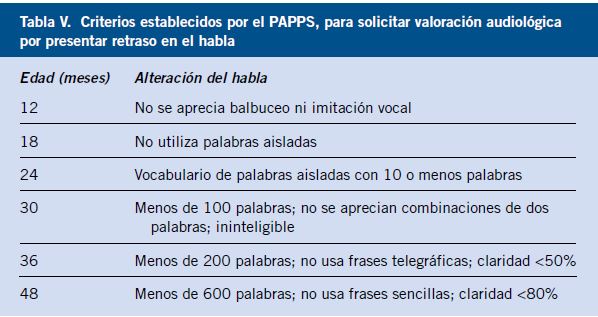
Los traumatismos en los niños pequeños se producen en casa, con los juguetes o en la cocina (con aceite, agua hirviendo, productos de limpieza, utensilios de cocina). Los más pequeños, si sufren la mordedura de un perro (Fig. 1), aunque esté familiarizado con él, las lesiones oculares ocurren en un 15% de los casos.
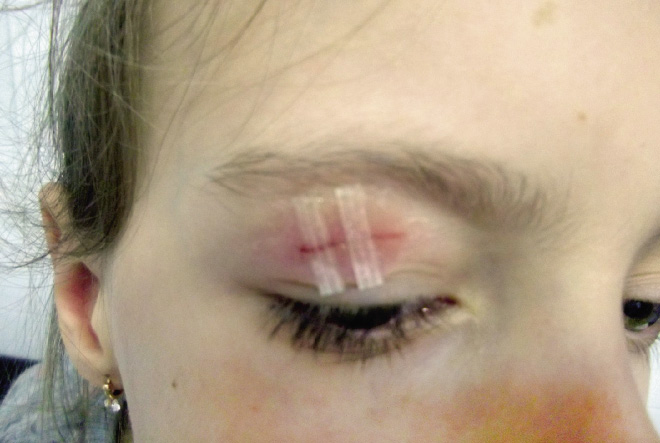
Figura 1. Mordedura de perro.
En niños mayores de 8 años con los deportes, una de las causas más importantes (pelota, tirachinas, palos, petardos, piedras, clips de papel, cuerdas elásticas, proyectiles, dardos, arcos, flechas y balines de aire comprimido –los balines causan lesiones especialmente devastadoras–). Los varones de 11 a 15 años son los más vulnerables(3). No podemos olvidar deportes como el hockey, deportes con raqueta, béisbol, baloncesto, esgrima, squash, boxeo, artes marciales de contacto, lucha libre y, actualmente, el paint-ball (juego con balas de pintura –juego de guerra–). Las lesiones por balas de pintura son actualmente el traumatismo ocular más frecuente y más grave relacionado con la práctica del deporte. El diámetro del paint-ball es inferior a 2 cm y alcanza velocidades de 90 a 150 metros por segundo. En los últimos años, la protección ocular en algunos de estos deportes ha reducido el porcentaje de lesiones(4).
Se han descrito también lesiones oculares por sistemas de airbag (quemaduras, laceraciones, abrasión corneal, hipema, desprendimiento de retina e, incluso, estallido ocular), aunque el efecto protector frente a un accidente mortal tiene mayor peso que el pequeño riesgo asociado.
La mayoría de lesiones oculares son evitables con los sistemas de protección actuales disponibles. Los pacientes con ambliopía deben ser identificados y deben tener especial cuidado si practican algún deporte de riesgo, pues la lesión en el ojo útil mermaría su calidad de vida. En la página web de la academia americana de oftalmología, se habla de prevención de lesiones oculares: www.AAO.org (Preventing Eye Injuries Children).
Historia clínica y exploración general
Realizar una buena historia clínica es muy importante(5). Al tratarse de un niño, la mayor parte de la información la obtendremos de los cuidadores (frecuentemente los padres) y no es tarea fácil. Las circunstancias del traumatismo deben ser recordadas cuidadosamente, ya que pueden tener implicaciones médico-legales importantes.
Debe interrogarse el cuándo, cómo y con qué se ha producido la lesión(6).
Hay que preguntar al paciente, si ello es posible por la edad, por cambios visuales graduales o repentinos desde que sufrió el trauma o síntomas, como dolor, diplopía y fotofobia.
Anotar el momento y lugar del accidente, el mecanismo de la lesión, si ha sido accidental o intencional, la presencia de testigos, el objeto que causa el accidente, la sospecha y el tipo de cuerpo extraño, si ha habido lesiones químicas por álcalis, si ha habido lesión por animal, tipo de animal y si ha sido espontáneo o provocado. También es muy importante averiguar si hay enfermedades oculares preexistentes y el nivel de agudeza visual del paciente antes del accidente (si es posible).
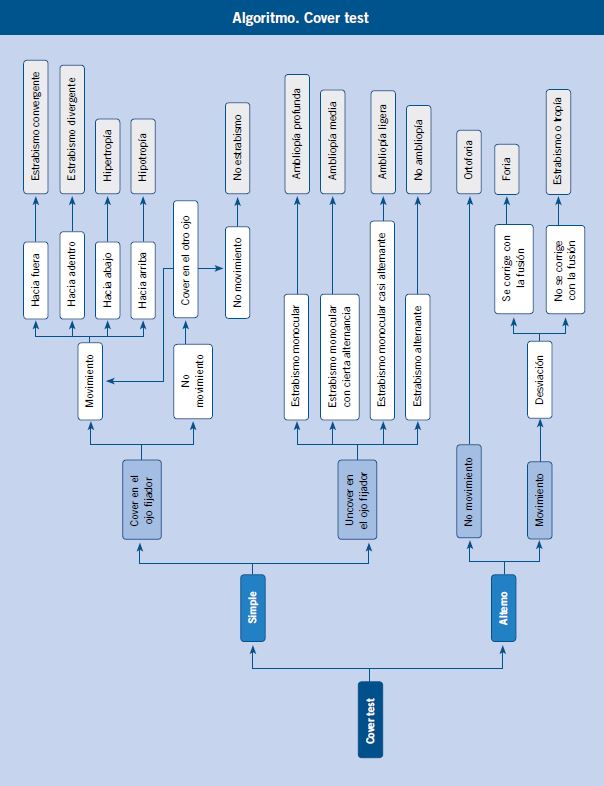
La exploración no es sencilla. Debe ser realizada por personal cualificado. La ansiedad y el dolor del paciente dificultan la cooperación. Si es difícil explorar al niño, deberá efectuarse bajo sedación o anestesia general.
Un dato muy importante y por el que siempre debemos comenzar la exploración es la Agudeza Visual (AV). Hay que explorar cada ojo por separado. Si el niño es mayor de 3 años, se debe realizar mediante la escala E de Snellen. Valorarla en menores de 3 años resulta difícil: hay que enseñarle un juguete con colores vivos, prestando atención a la capacidad del niño de fijar la vista en ese objeto. Puede ser que, en ese momento, se intente quitar el parche de su ojo sano, lo cual nos hará sospechar una menor AV en el ojo afectado.
Si la edad lo permite, valorar la visión cromática (el color rojo puede parecer gris si hay disfunción del nervio óptico). Es importante para el diagnóstico, pronóstico y evaluación médico-legal.
Las pupilas nos dan información del sistema visual y de una posible patología intracraneal. Si el paciente está inconsciente, es la única forma de información. Explorar los reflejos fotomotores directo y consensuado es muy importante, así como la forma de la pupila en el ojo traumatizado. Por ejemplo, la presencia de un defecto pupilar aferente relativo (pupila que se dilata paradójicamente al ser iluminada, tras haber iluminado previamente el ojo contralateral) nos indica una posible lesión en la vía aferente visual, ya sea a nivel del nervio óptico (alteración más frecuente), o a nivel retiniano (lesión que debe ser muy extensa para provocar dicha alteración pupilar).
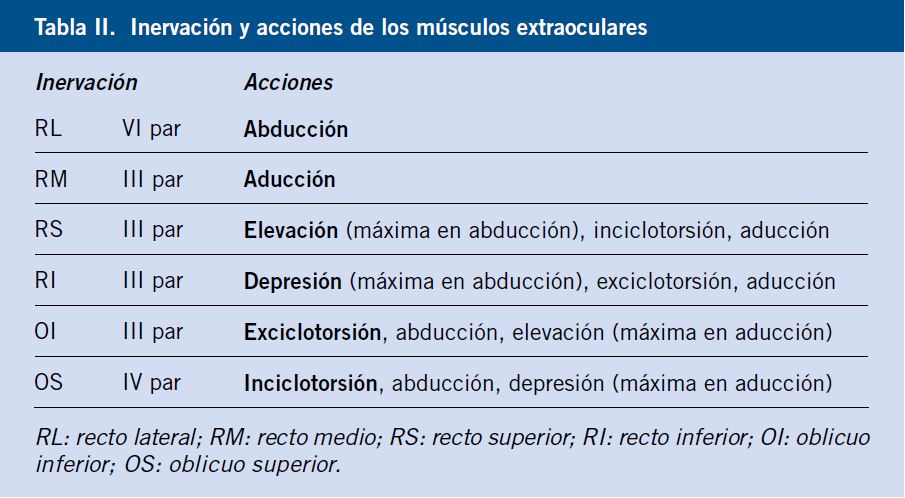
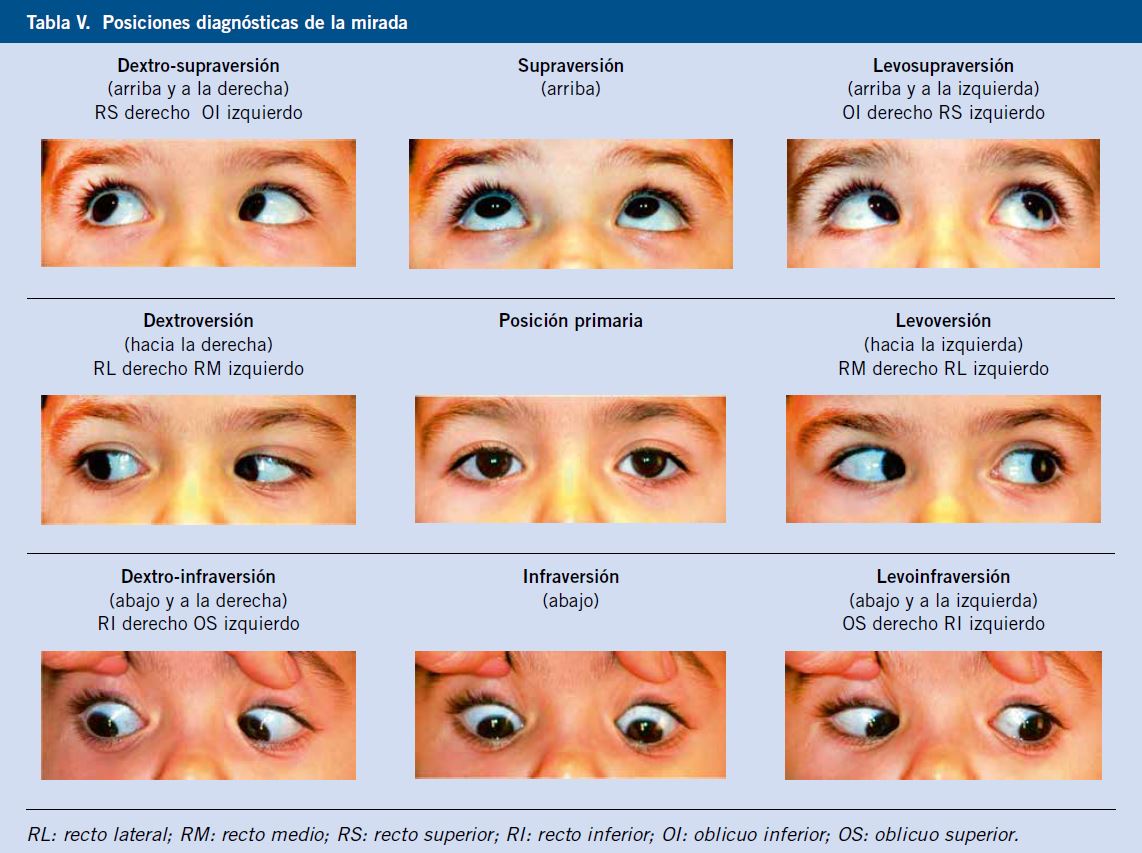
Valorar la motilidad extraocular es importante ante la sospecha de lesión de la órbita o de un nervio craneal.
La presión intraocular es otro dato importante. Siempre, antes de valorarla, debemos asegurarnos de que no existe un traumatismo ocular abierto, ante lo cual está totalmente contraindicado tomarla. La toma se realiza mediante una ligera presión con ambos dedos índices sobre el párpado superior, ejerciendo presión de manera alterna y después comparando con el ojo contralateral. Suele ser complicado detectar ligeras variaciones, por lo que únicamente nos servirá para descartar grandes hipertonías. Ante un aumento de presión importante, debemos remitir inmediatamente al oftalmólogo para su diagnóstico y tratamiento.
La sospecha de abuso del niño debería ser considerada si la historia es inconsistente con la lesión ocular del paciente: historia del cuidador contradictoria o múltiples visitas al servicio de urgencias. La exploración oftalmológica puede ayudar en el diagnóstico del síndrome del bebé zarandeado (hemorragias intracraneales e intraoculares –intra, pre o subretinianas– sin signos de traumatismo externo).
La exploración externa del paciente: observar la cabeza, cara, región periorbitaria y párpados. Anotar equimosis, edema, ptosis, laceraciones y cuerpos extraños, enoftalmos y exoftalmos. La región periorbitaria debe palparse para descartar crepitación o deformidades en huesos orbitarios. La hipoestesia infraorbitaria, crepitación, enoftalmos y deformidad orbitaria pueden indicar fractura orbitaria por compresión (blow-out).
Respecto a las exploraciones complementarias, la radiografía simple tiene menos valor que las exploraciones que se efectúan actualmente, como el TAC y la Resonancia Magnética (RM). Sin embargo, es útil para realizar la primera valoración rápida o para orientarnos si no disponemos de las otras exploraciones complementarias de urgencias. Con ella, podemos valorar la presencia de un cuerpo extraño intraorbitario o intraocular, o la fractura de las paredes de la órbita. No detecta cuerpos de plástico o madera por ser radiolúcidos y tampoco se visualizan las partes blandas.
El TAC orbitario es la prueba que más información nos aporta, localización exacta de cuerpos extraños y fracturas, nos da imágenes del globo ocular, músculos y paredes orbitarias. Siempre se debe realizar ante la sospecha de perforación ocular. Aunque, al ser una prueba más específica, debe ser valorada siempre por un radiólogo y un oftalmólogo.
La RM visualiza mejor las lesiones vasculares, patología intracraneal, trombosis del seno cavernoso y enfermedades desmielinizantes e inflamatorias del nervio óptico. No suele ser tan útil para localizar lesiones de traumatismos oculares, y no debe ser la primera prueba diagnóstica. Siempre hay que descartar antes, mediante otras pruebas de imagen, la ausencia de cuerpos extraños metálicos.
La ecografía es útil, pero no debe realizarse si se sospecha perforación ocular. Proporciona la mayor resolución e información anatómica del segmento posterior ocular. Puede visualizar cuerpos extraños intraoculares, radiopacos y radiolúcidos y patología del vítreo, retina, coroides y esclerótica. También la glándula lagrimal, músculos, nervio óptico y tejidos blandos de la órbita.
Biomicroscopía o exploración del segmento anterior ocular
Párpados, conjuntiva, córnea y esclera
La mayor parte de las lesiones de la superficie ocular son benignas y se resuelven sin tratamiento, sin embargo, requieren una exploración meticulosa (incluso en lesiones que parecen de poca importancia) y un tratamiento precoz para evitar secuelas permanentes y complicaciones tardías.
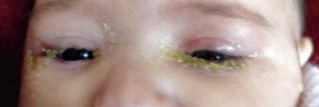
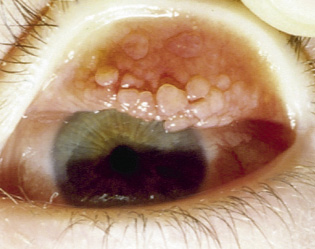
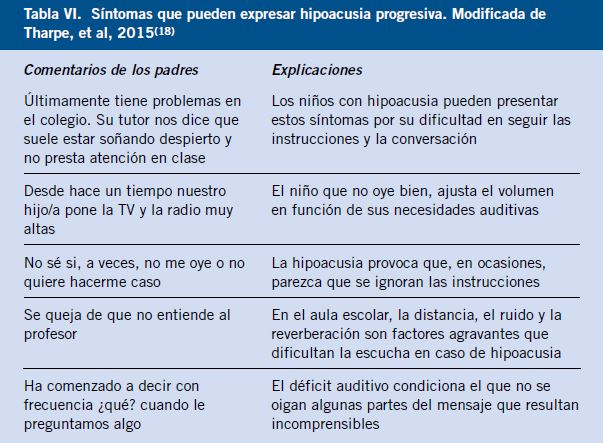
Párpado (Figs. 1-4)
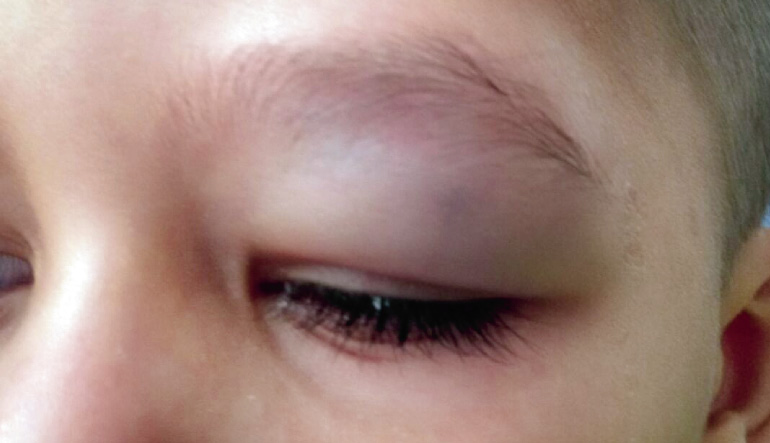
Figura 2. Traumatismo palpebral.
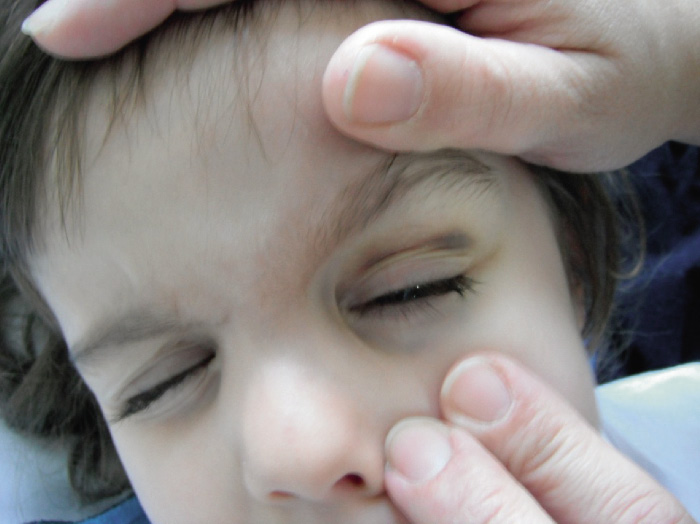
Figura 3. Traumatismo palpebral por pelota de tenis.

Figura 4. Herida a nivel del párpado superior e inferior, puede iniciar una celulitis preseptal.
La equimosis (hematoma u ojo morado) aparece en el párpado tras una contusión y se reabsorbe espontáneamente. Por efecto de la gravedad, puede extenderse al párpado inferior, lo cual debe advertirse a las familias. Puede tratarse con compresas frías y analgésicos.
Cuando hay una herida en el párpado debe tenerse en cuenta la localización porque:
• Si la herida afecta al párpado superior, puede estar comprometido el músculo elevador; la sutura incorrecta daría lugar a ptosis palpebral. Frecuentemente, hay salida de grasa orbitaria por la herida. No debe dejarse curar por segunda intención.
• Si la herida afecta al borde palpebral, la sutura correcta se hará alineando los bordes de la herida para evitar eversión o inversión del párpado y evitar la aparición de epífora, irritación o defecto de humidificación corneal.
• Cuando la herida afecta a la zona interna del párpado, puede dañar el conducto lagrimal, requiriendo una reparación con microcirugía por parte del oftalmólogo.
• También puede dañarse la glándula lagrimal situada en la zona externa del párpado.
Siempre, ante una herida en el párpado, remitir al oftalmólogo para su sutura. Una malposición palpebral o no tener en cuenta la vía lagrimal a la hora de realizar la sutura nos puede dar complicaciones oculares a posteriori.
La infección de la herida palpebral secundaria al traumatismo puede complicarse con una celulitis periorbitaria.
La ptosis palpebral puede deberse al hematoma y a la reacción inflamatoria, que desaparece en unos días, o bien a la lesión del músculo elevador del párpado, en cuyo caso el pronóstico es peor.
El hematoma palpebral bilateral en anteojos o también llamado de “ojos de panda” debe hacernos pensar en la fractura de la base del cráneo.
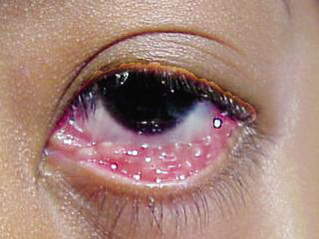
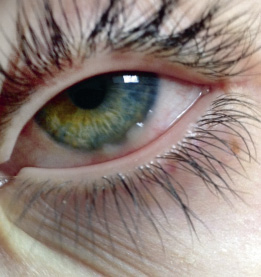
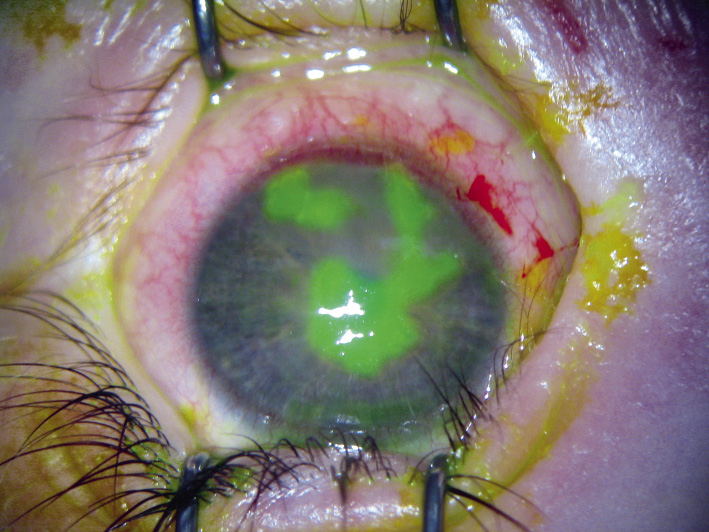
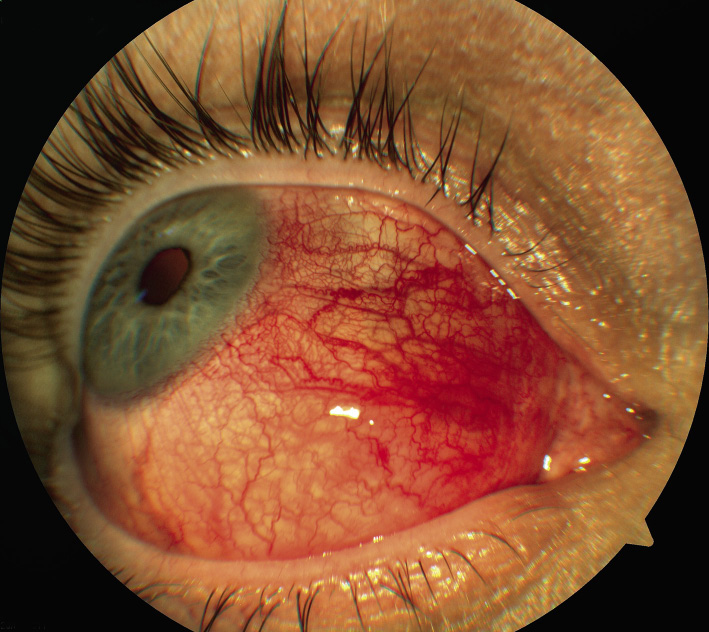
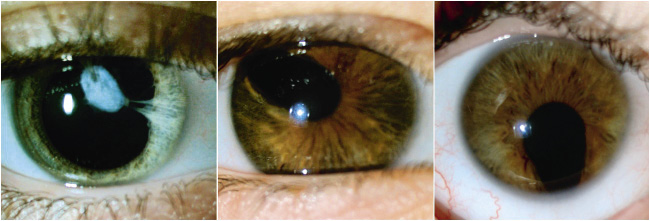
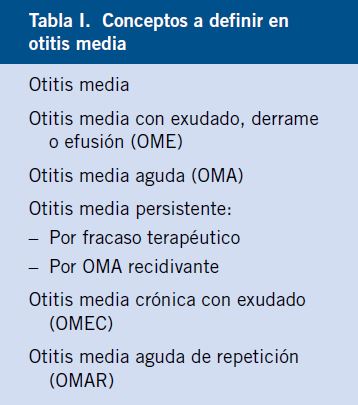
Conjuntiva y córnea (Figs. 5-8)

Figura 5. Traumatismo con un lápiz: erosión corneal.

Figura 6. Erosión corneal con un papel.

Figura 7. Cuerpo extraño corneal.
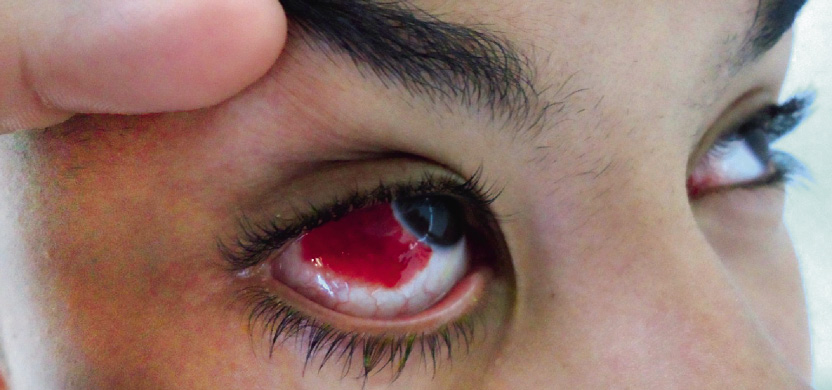
Figura 8. Hiposfagma o hemorragia subconjuntival.
Cuando exploramos la conjuntiva o la córnea, tras un traumatismo, podemos encontrarnos con: hemorragia subconjuntival (hiposfagma), erosión de la superficie, quemosis, enfisema, abrasiones, laceraciones y cuerpo extraño conjuntival (lentes de contacto) o cuerpo extraño corneal y abrasión corneal.
La hemorragia subconjuntival traumática tiene un aspecto llamativo. La espontánea suele deberse a una maniobra de Valsalva que aumenta la presión venosa (tos: tosferina, estornudo, vómitos, levantar objetos pesados). Las conjuntivitis víricas o bacterianas también son una causa frecuente. El frotamiento ocular enérgico, también puede producirla. Desaparece en unos días, pero puede durar semanas. La hemorragia subconjuntival debe ser limitada a la zona anterior. La mayoría de ellas son benignas. Si es extensa (ocupando toda la conjuntiva y fondos de saco conjuntival) debería pensarse en la posibilidad de fractura del techo o suelo de la órbita e incluso hemorragia intraocular o rotura del globo.
La sensación de arenilla o cuerpo extraño es la molestia más frecuente en estas urgencias oftalmológicas.
La lesión conjuntival da lugar a un leve dolor por la escasez de inervación sensorial.
Tras una lesión corneal, la capa basal del epitelio corneal y los nervios corneales superficiales quedan expuestos a estímulos externos, siendo el dolor más intenso.
Clínicamente, suele aparecer: dolor, lagrimeo, fotofobia, enrojecimiento ocular, blefaroespasmo y disminución de la visión. Los movimientos del globo ocular y el parpadeo aumentan el dolor y la sensación de cuerpo extraño.
Para visualizar las lesiones hay que aplicar fluoresceína. La fluoresceína tiñe las lesiones en el epitelio, ya sean conjuntivales o corneales. Será la exploración diagnóstica principal. Utilizar tiras estériles de papel o colirio, en el ojo afectado, e iluminarlo con luz azul de cobalto y podremos apreciar las erosiones en color amarillo verdoso. La tira de fluoresceína deberá estar ligeramente humedecida con colirio anestésico doble, lo que nos ayudará a realizar mejor la exploración. El paciente deberá mirar hacia arriba, mientras el extremo de la tira contacta con el fondo de saco inferior, evitando el roce con la córnea.
Las erosiones corneales curan rápidamente, pero pueden complicarse con recurrencias; reaparece la lesión al cabo de un tiempo sin haber sufrido ningún traumatismo, sobre todo, al levantarse por las mañanas, lo que se denomina síndrome de erosión corneal recidivante. Esto se produce porque las uniones hemidesmosómicas de las nuevas células epiteliales corneales son débiles, al estar un tiempo prolongado con los párpados cerrados al dormir, el epitelio nuevo se engancha literalmente al párpado superior, produciéndose una nueva erosión corneal al abrir el párpado de forma espontánea.
La morfología de las erosiones corneales nos puede ayudar a la hora de realizar el diagnóstico. Por ejemplo, las erosiones lineales nos hacen sospechar cuerpos extraños subtarsales. La eversión del párpado superior y la observación de los fondos de saco conjuntivales son técnicas útiles para localizar y eliminar los cuerpos extraños. Ante una erosión corneal, siempre debemos evertir el párpado superior en busca de cuerpos extraños.
El pediatra puede intentar retirar el cuerpo extraño, instilando previamente un colirio anestésico, mediante irrigación con suero con un chorro fuerte, con algodón o con una gasa humedecida; debe revertirse el párpado superior para detectar cuerpos extraños en el fondo de saco, en la zona subtarsal, para lo cual el paciente debe mirar hacia abajo. Hay que evitar los instrumentos afilados.
Cuando el cuerpo extraño es tierra o serrín, el lavado abundante con suero fisiológico será útil, pero ante la mínima dificultad enviar el paciente al oftalmólogo. En ocasiones, el niño deberá ser sedado para la retirada del cuerpo extraño en el quirófano.
Pese a la creencia extendida, tras la extracción de un cuerpo extraño, o bien tras una erosión corneal o conjuntival traumática, nunca debemos ocluir el ojo. Únicamente pautar pomada antibiótica y colirio midriático (ciclopléjico) para disminuir el dolor mediante la relajación del músculo ciliar. Tras el diagnóstico de una erosión conjuntival o corneal, o tras la extracción de un cuerpo extraño, siempre debemos remitir al paciente al oftalmólogo para su posterior seguimiento. Aunque la mayoría de lesiones de este tipo curan en unas horas o en un par de días, debemos asegurarnos que es visitado por un especialista, para evitar posibles complicaciones.
La oclusión queda reservada para las erosiones de origen fototraumático o por radiación (erosiones estériles). La oclusión debe realizarse con los párpados cerrados, con dos gasas dobladas y con ligera presión impidiendo así el movimiento palpebral y mantenerlo durante 24 horas. Deberán utilizarse pomadas que tienen un efecto lubricante, aunque enturbien la vista. Hay que aconsejar que no se froten los ojos. Los antibióticos aminoglucósidos, producen un efecto inhibitorio sobre la epitelización. Los macrólidos son una buena opción para el tratamiento (pomada de eritromicina, por ejemplo). No debe permitirse que el paciente utilice gotas anestésicas tópicas para el dolor, que impiden la emigración epitelial normal y favorecen que la abrasión corneal aumente de tamaño y el daño corneal se haga permanente. Si el dolor es intenso, el paciente deberá tomar analgésicos por vía oral. Los defectos corneales traumáticos, aunque sean moderadamente grandes, suelen cicatrizar en uno o dos días sin parches en los niños pequeños.
Como secuelas tardías de laceración corneal profunda, podemos tener el astigmatismo y cicatrices corneales (leucomas) que pueden reducir la agudeza visual.
Los cuerpos extraños metálicos en la superficie corneal empiezan a oxidarse a las pocas horas, son tóxicos y deben ser eliminados. Con los cuerpos extraños profundos de material inerte (plástico o cristal), se puede mantener una actitud expectante, pues con el tiempo migran en dirección anterior y se extraen con más seguridad. Ante la presencia de uno de estos tipos de cuerpos extraños, remitir siempre al oftalmólogo para su tratamiento y posterior seguimiento.
Quemaduras y causticaciones
Las causticaciones por ácidos y álcalis son urgencias que requieren un tratamiento inmediato antes del diagnóstico de las lesiones. El tratamiento consiste en la irrigación del ojo con 500 cc de suero fisiológico o Ringer lactato.
La cantidad de daño tisular se relaciona directamente con la duración del contacto entre el agente químico y el ojo. “Cada segundo cuenta” en una lesión química. La irrigación inmediata es vital.
Las quemaduras y causticaciones son una verdadera urgencia oftalmológica: en las quemaduras térmicas, los párpados y la córnea son las zonas más afectadas. El agua y el aceite no suelen provocar lesiones tan graves como los agentes químicos(7). También la pólvora de los petardos provoca quemaduras. Las quemaduras químicas en la infancia suelen deberse a detergentes o disolventes orgánicos que se encuentran en productos de limpieza domésticos.
Los álcalis suelen penetrar el ojo con mayor facilidad que los ácidos. Su morbilidad es significativamente mayor. Las causticaciones oculares pueden lesionar el epitelio corneal y conjuntival, la membrana basal, el estroma corneal, el endotelio de los vasos conjuntivales y la epiesclera.
Los álcalis que pueden afectar el globo ocular son: el amoniaco, el hidróxido sódico (lejía, desatascadores), la potasa cáustica, el hidróxido de magnesio y el hidróxido cálcico (cemento, yeso y cal). Los ácidos son el sulfúrico (batería del automóvil), el ácido clorhídrico (provoca lagrimeo a distancia), el ácido nítrico (con un contacto prolongado se comporta como el clorhídrico) y el ácido acético (precisa de largo tiempo de contacto y una concentración superior al 10%).
El álcali provoca lesiones más graves, ya que penetra rápidamente en el interior del ojo, saponifica los ácidos grasos de las membranas celulares epiteliales, desnaturaliza el colágeno y produce trombosis vascular. Ante una causticación por un álcali, antes de empezar la irrigación aplicar unas gotas de aceite de oliva sobre la superficie ocular, ayuda a detener la reacción saponificadora que se produce. Los ácidos provocan menos daño porque el ion hidrógeno precipita las proteínas evitando la penetración del producto a través de la córnea.
El pronóstico, por tanto, dependerá de si el agente es un álcali o un ácido, del área de exposición y de la concentración de la sustancia química y del tiempo de contacto con el tejido del ojo.
Esta urgencia es la única en la cual iniciamos el tratamiento antes que la exploración, con irrigación abundante (las 3 íes de irrigación, irrigación, irrigación) con 500 cc de suero fisiológico o Ringer lactato. No debe efectuarse presión sobre el globo ocular y hay que tener en cuenta que, a mayor espacio de tiempo entre la producción de la lesión y el lavado, el pronóstico será peor.
Hay que efectuar la limpieza, con una gasa, de los restos que pueden estar en contacto con la conjuntiva y con los fondos de saco conjuntival superior e inferior. Deben administrarse analgésicos por vía oral en las primeras horas tras el accidente. La quemadura corneal con cigarrillos es la lesión térmica más frecuente de la superficie ocular del niño. Son accidentales y no hay que pensar en manifestación de maltrato. Es debido a que los ojos del niño están a la altura del cigarrillo que tiene el adulto. Estas quemaduras suelen curar con rapidez y sin cicatrices.
La ceniza del cigarrillo se comporta como un álcali al cual se añade el efecto del calor.
Ante una causticación debe remitirse el paciente al oftalmólogo, que determinará su gravedad e iniciará el tratamiento pertinente. La mayoría se resuelven con: pomada antibiótica, colirio con corticoide, colirio ciclopléjico y vitamina C vía oral. Las más graves pueden precisar incluso de tratamiento quirúrgico. A consecuencia de estas lesiones, se pueden formar cicatrices, glaucoma, cataratas y precisar un trasplante corneal.
Otro agente químico son los pegamentos, que si se han adherido a los párpados, habrá que recortar las pestañas, ya que producen adherencias muy firmes, y despegar los bordes con ligeras tracciones. El pegamento sobre la superficie ocular está poco adherido y al traccionarlo sale como un bloque.
Los petardos
El 5% de los traumatismos oculares son consecuencia de los fuegos artificiales, tan tradicionales en nuestro país. Las 2/3 partes de los accidentes relacionados con petardos se producen en los domicilios particulares. Se afecta tanto el que observa como el que lanza el artefacto. Se aconseja siempre la supervisión de los padres ante la manipulación de este tipo de artefactos por parte de niños. También es recomendable el uso de gafas de protección. Incluso las bengalas que parecen inofensivas, pueden provocar quemaduras importantes.
Lesiones fototraumáticas y por radiaciones
Las radiaciones ultravioletas son la causa más frecuente de lesiones tras la exposición solar en la playa o en la nieve.
Las lesiones suelen ser bilaterales y pueden manifestarse en forma de un punteado que tiñe con fluoresceína el área interpalpebral, hasta zonas extensas de desepitelización. Los síntomas (intenso dolor) aparecen de 8 a 12 horas tras la exposición y no hay relación entre el tiempo de exposición y la intensidad de las manifestaciones clínicas.
La retinopatía solar es una lesión producida por la luz del sol en personas que observan un eclipse solar. Aparece una pequeña quemadura en la retina que no ocupa toda la superficie, dando lugar a una disminución de la visión.
Los punteros láser de uso doméstico, aunque podrían producir fotocoagulación de la retina-lesión directa de la fóvea- (mirando fijamente más de 10 segundos al puntero), no suelen causar afectación, pues el tiempo de exposición es inferior a 1 segundo.
Lesiones del globo ocular con afectación de la cámara anterior
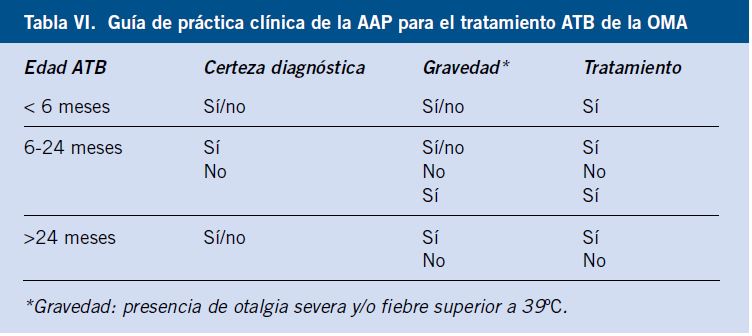
Hipema (Fig. 9)
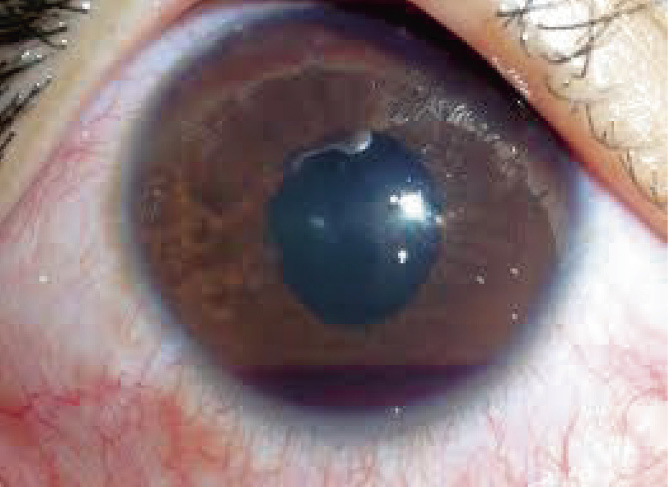
Figura 9. Hipema.
Consiste en la acumulación o presencia de sangre en la cámara anterior del ojo (en la parte más inferior) y se visualiza como un nivel rojo entre la córnea y el iris. Es una verdadera urgencia oftalmológica. Puede ser reconocido con la luz de la linterna.
El hipema es secundario a traumatismos con objetos de tamaño más pequeño que el reborde de la órbita y consecuente lesión de los vasos sanguíneos de la raíz del iris o del cuerpo ciliar. Ante la aparición de un hipema, hay que descartar la rotura o estallido ocular y valorar el estado del iris y el cristalino(8).
Puede acompañarse de lesiones en el iris, cuerpo ciliar y cristalino. En pacientes de origen africano y con antecedentes de hemoglobinopatía falciforme o drepanocitosis, debe descartarse esta enfermedad. Debe evitarse que vuelva a sangrar en los días posteriores al traumatismo.
Existe bastante controversia en referencia a su tratamiento, algunos autores postulan por un tratamiento ambulatorio, mientras que otros siguen defendiendo el ingreso hospitalario. Actualmente, el ingreso se reserva para niños pequeños (difícil que sigan un reposo estricto en casa) o pacientes con hemoglobinopatías. Los esteroides son el pilar del tratamiento de los hipemas. El reposo en cama, con la cabeza elevada (posición de la cama en 45º), protector ocular (para evitar tocarse) y sedación en algunos casos. Deben ser controlados diariamente por el oftalmólogo para conocer la tensión intraocular y detectar los cambios que se produzcan con el hipema.
La hematocórnea es la impregnación de la córnea por el pigmento férrico de la sangre. Se produce en hipemas masivos. En niños puede ser necesario un trasplante corneal. El glaucoma puede ser una complicación secundaria al hipema. Ante el diagnóstico de una hematocórnea, derivar al paciente al oftalmólogo de forma urgente, la mayoría de veces es necesario el drenaje del sangrado de forma quirúrgica.
La tinción hemática corneal puede tardar meses o años en desaparecer y puede provocar en la infancia ambliopía por deprivación, con las consecuencias sobre la función visual. Por eso debe tratarse con la mayor rapidez posible.
Si el sangrado afecta a más del 50% de la cámara anterior, requerirá intervención quirúrgica para evacuar el coágulo e irrigar la cámara anterior.
Clínicamente, hay dolor ocular y disminución de la visión o visión borrosa.
La somnolencia es frecuente en los niños y, a veces, es tan intensa que obliga a realizar un examen neurológico, sobre todo, si ha habido un traumatismo craneal.
Si no hay antecedente de traumatismo previo, deberá descartarse como desencadenante una enfermedad hematológica (trastorno de coagulación, leucemia), retinoblastoma, xantogranuloma juvenil y la ya mencionada drepanocitosis.
El hipema secundario a un traumatismo con un objeto pequeño es una verdadera urgencia que debe ser valorada por el oftalmólogo.
Iris y cuerpo ciliar
La característica de la afectación del iris y cuerpo ciliar es la fotofobia. A menudo, hay una inyección de los vasos sanguíneos episclerales o conjuntivales alrededor del limbo, denominándose hiperemia conjuntival ciliar. La visión generalmente está reducida y la pupila puede ser más pequeña en el ojo afecto respecto al ojo sano. La lesión del músculo esfínter del iris dará lugar a una midriasis.
Clínicamente hay dolor, fotofobia, anisocoria, inyección conjuntival perilimbal (hiperemia ciliar) y visión borrosa. También puede aparecer dificultad para el enfoque de cerca, por espasmo o parálisis de la acomodación, y dificultad en la constricción y dilatación pupilar tras la estimulación luminosa.
Si, tras el traumatismo ocular, hay rotura del esfínter iridiano, aparece la discoria o pupila irregular, pupila en forma de D. La afectación del iris suele acompañarse de hipema, por lo que debe valorarlo un oftalmólogo con urgencia.
Las lesiones de la cámara anterior son una de las secuelas más frecuentes de los traumatismos oculares contusos.
Cristalino
Evaluar el cristalino es fundamental en la exploración del ojo que ha sufrido un traumatismo ocular. El traumatismo es la causa más frecuente de la luxación o subluxación del cristalino(7).
La catarata y la luxación o subluxación que aparecen después de un traumatismo ocular indican que este ha sido importante (Fig. 10). Provoca un cuadro de pérdida de visión importante de aparición brusca. También suele acompañarse de hipema y hay que derivarlo al oftalmólogo.
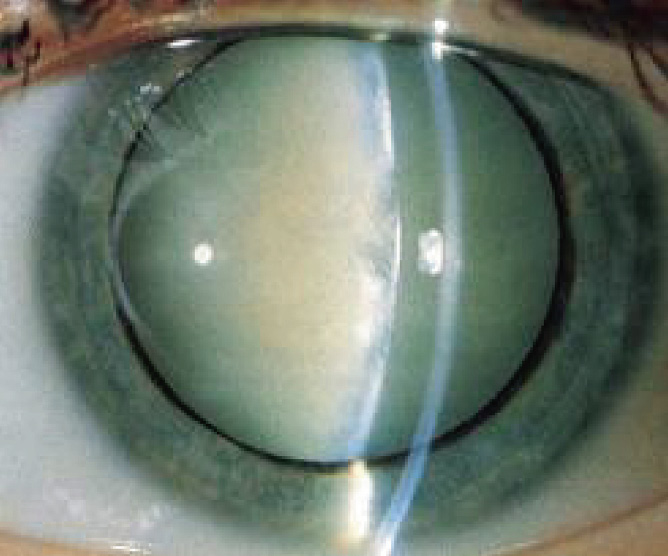
Figura 10. Catarata traumática.
Los traumatismos contusos pueden producir cataratas de aparición inmediata o incluso años después.
Si la catarata da lugar a pérdida de agudeza visual y el paciente es menor de 8 años, hay que plantear una intervención quirúrgica precoz para evitar la ambliopía. El glaucoma y el contacto corneal secundarios a la luxación del cristalino también obligarían a plantear la intervención quirúrgica.
La luxación del cristalino frecuentemente es de origen traumático en la infancia. Nos indicará que el trauma ha sido importante. Ante una luxación o subluxación del cristalino, en ausencia de traumatismo ocular, valorar la posibilidad de un síndrome de Marfan.
Lesiones del globo ocular con afectación del segmento posterior: vítreo, retina y coroides
La gravedad de las lesiones en las estructuras del segmento posterior es variable: oscila entre las contusiones retinianas periféricas, que se pueden resolver espontáneamente, hasta el desprendimiento de la retina, que puede poner en peligro la visión.
Si el paciente no presenta una transparencia de medios para poder realizar un fondo de ojo, deberá solicitarse una ecografía para examinar el segmento posterior. La ecografía permitirá detectar: desgarros ocultos de la retina, desprendimiento de retina, hemorragias subretinianas, hemorragia subcoroidea (desprendimiento coroideo), desprendimiento del vítreo posterior, luxación del cristalino, roturas ocultas del globo ocular y cuerpos extraños intraoculares.
Hemorragia vítrea
Sangre en la cavidad vítrea. Es la complicación del polo posterior más frecuente tras el traumatismo ocular. Son síntomas las moscas volantes y suele haber pérdida visual súbita y grave. Algunos pacientes dicen que ven “en rojo”. La presencia de hemorragia vítrea en lactantes o niños debe alertar sobre la posibilidad de malos tratos. Los niños en edad de ambliopía son un reto terapéutico. En los lactantes puede aparecer ambliopía por deprivación las primeras semanas de la hemorragia vítrea. Pueden producirse anisometropías de más de 10 dioptrías. También puede aparecer como complicación un glaucoma.
Contusión, edema, hemorragia y desprendimiento de retina
Puede producirse tras lesiones de golpe o contragolpe (compresión y expansión del globo ocular). En una contusión retiniana aparecen áreas geográficas de retina blanqueada. Si la contusión afecta a la mácula, (llamado edema de Berlin), se puede producir pérdida de visión aguda. Si se afecta la periferia de la retina, el paciente visualmente está asintomático. Si se afecta la fóvea (área central macular), la pérdida de visión puede ser permanente.
Rotura coroidea
Complicación muy grave. Si aparece nos indica que el traumatismo ocular ha sido importante. Hay afectación de la mácula en más de la mitad de las roturas coroideas. La afectación macular (edema, hemorragia o desprendimiento) comporta la pérdida de visión inmediata. La mayoría de veces no se recupera la agudeza visual tras la reabsorción de las hemorragias y el edema secundario a la rotura. La afectación macular va emparejada con una pérdida de agudeza visual permanente.
Hemorragia subcoroidea (desprendimiento coroideo)
Suele ser una lesión rara, pero grave. Aparece tras traumatismos contusos mediante el mecanismo de compresión y expansión del globo ocular. La sangre se acumula en el interior del espacio virtual entre la coroides y la esclerótica. Puede estar asociada a un estallido ocular oculto. Puede resolverse espontáneamente o aparecer complicaciones tardías, como el desprendimiento de retina.
Fractura de la órbita (Fig. 11)
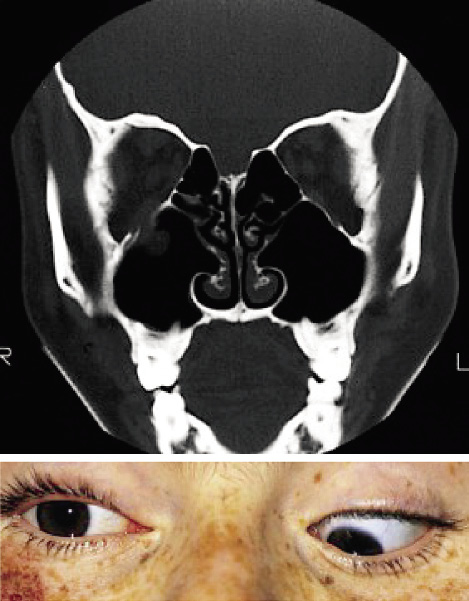
Figura 11. Fractura Blowout de la órbita derecha. Atrapamiento del músculo recto inferior.
El signo más evidente de la fractura del suelo de la órbita es la limitación de la mirada vertical.
El suelo de la órbita está formado por los huesos maxilar superior, cigomático y palatino. Tiene un grosor de 0,5 a 1 mm. La pared medial está formada por el hueso maxilar superior, el lagrimal y etmoides. La pared medial del hueso etmoides es muy delgada (0,2 a 0,4 mm) y se denomina lámina papirácea. La mayor parte de las fracturas de la pared medial se producen en él. El techo está constituido por el hueso frontal y el ala menor del esfenoides. La pared lateral externa la forman el ala mayor del esfenoides y las apófisis orbitarias del cigomático y del hueso frontal.
Los accidentes con vehículos a motor, los puñetazos en los ojos y las lesiones deportivas con impacto secundario a pelotas son las causas más frecuentes de traumatismos en la órbita.
Hay lesiones contusas y penetrantes. Los puños, pelotas de tenis, golf, squash, bates de béisbol, botellas o superficies romas, como el salpicadero del coche o las puertas, pueden provocar lesiones contusas.
Las paredes orbitarias pueden fracturarse y el globo ocular puede perforarse, incluso sin que el objeto haya penetrado directamente en los tejidos(9).
Los objetos penetrantes suelen ser de tamaño inferior al de la órbita (cuchillos, lapiceros, ramitas, balas, clavos y la punta del paraguas). La velocidad del objeto es más importante que el tamaño. Los objetos proyectados a gran velocidad transmiten más energía cinética y, por tanto, ocasionan mayor daño tisular.
El techo y la pared lateral externa son más gruesas y menos propensas a las fracturas.
Las fracturas más frecuentes se localizan en el suelo de la órbita y la pared medial o interna, debido a que son las más débiles. La pared externa y la pared superior son más resistentes y se fracturan en traumas muy intensos, pero en los niños menores de 7 años, estas fracturas del techo de la órbita se producen por falta de neumatización de los senos frontales y por la desproporción cráneo-facial.
El signo más evidente de la fractura del suelo de la órbita es la diplopía o la imposibilidad de llevar la mirada hacia arriba.
Lesiones orbitarias graves, pueden tener un aspecto externo trivial. Hay que investigar la disminución de la agudeza visual y la diplopía. Puede detectarse: equimosis, epistaxis ipsilateral, enfisema orbitario, sugestivo de lesión de la pared medial (debido al paso de aire desde el seno etmoidal a través de una fisura en la lámina papirácea) o inferior, hipoestesia de la mejilla y del labio superior, si hay afectación del nervio infraorbitario, y escalón en el reborde orbitario, en la fractura del suelo de la órbita cuando hay fractura directa(10).
La limitación de la motilidad ocular extrínseca, enoftalmos y diplopía en la mirada de frente y en la mirada inferior (para la lectura y para caminar) es indicativa de un tratamiento quirúrgico precoz. La no existencia de estos signos dará lugar a la abstención de la cirugía.
La presencia de exoftalmos sugiere que el volumen orbitario está incrementado por edema, hemorragia, aire o fragmentos óseos.
La secreción nasal de líquido trasparente puede indicar rinorrea de LCR, que nos alertará acerca de la fractura de la fosa craneal anterior.
La diplopía secundaria al traumatismo es debida a restricción muscular extraocular. Puede ser una limitación leve por el edema orbitario o puede ser debida a atrapamiento muscular (el más propenso es el músculo recto inferior que discurre a lo largo del suelo de la órbita) en el interior de la fractura, con lo cual precisará intervención para liberar el músculo y reconstruir el suelo de la órbita.
Debido a la fractura, puede producirse herniación del tejido orbitario al seno maxilar o etmoidal.
Hay que examinar la vía lagrimal, que estará afectada en un 20% de las fracturas de la pared medial.
La TAC orbitaria, con cortes axiales y coronales, y la radiología con proyección de Waters que muestra el suelo de la órbita y el seno maxilar, son las técnicas de imagen de elección para valorar a los pacientes con traumatismo orbitario. La imagen en gota suspendida en el seno maxilar superior es característica de las fracturas del suelo de la órbita.
No hay que olvidar explorar el globo ocular detalladamente, ya que se encuentran perforaciones y lesiones oculares asociadas a estos traumatismos.
En el recién nacido, tras un parto con fórceps, hay que valorar el traumatismo ocular obstétrico. Puede existir contusión ocular; por lo tanto, hay que explorar el globo ocular. En ocasiones, hay hematomas palpebrales, hemorragias subconjuntivales, edema corneal, lagoftalmos, blefaroptosis y hemorragia retiniana. La hemorragia macular ocurre en el 4% de recién nacidos (raramente dará lugar a ambliopía). En las primeras 4 a 6 semanas, las hemorragias se habrán reabsorbido sin secuelas.
El término “blow out fracture”, o fractura en estallido, se refiere a la expansión del volumen orbitario debido a la fractura de las paredes orbitarias delgadas hacia los senos paranasales adyacentes, sin compromiso del reborde. Estos huesos delgados son en realidad un mecanismo protector, ya que al romperse, reducen la presión sobre el globo ocular y previenen la rotura del ojo.
El 10-30% de las fracturas en estallido se acompañan de abrasiones corneales, hipema traumático, iritis, rotura del globo ocular, contusión retiniana, desprendimiento de retina y hemorragia retiniana.
Las fracturas de la pared medial habitualmente se asocian a fracturas del suelo de la órbita. Suelen comprometer el músculo recto medial.
Las fracturas del techo de la órbita aparecen en accidentes de automóvil y en caídas de una altura importante. Pueden afectar al seno frontal y, como complicaciones, mencionar la fuga de LCR, hemorragia intracraneal, limitación dolorosa de la mirada hacia arriba, ptosis, encefalocele traumático, meningitis y absceso cerebral.
Las fracturas de la pared lateral se deben a accidentes de automóvil, caídas o agresiones con objetos romos. Son muy poco frecuentes.
Lesión del nervio óptico
Como lesiones más importantes, encontramos la neuropatía óptica traumática y la, poco frecuente pero devastadora, avulsión del nervio óptico. Puede tratarse de una lesión directa, como la compresión o sección del nervio por un cuerpo extraño, como puede ser una bala. O lesiones indirectas debidas a un traumatismo craneal frontal, la rotación traumática del globo ocular y la hemorragia perineural.
En la neuropatía óptica traumática(6) hay alteración de la agudeza visual, alteración del test de Ishihara (colores afectados azul-amarillo) y defecto pupilar aferente relativo (pupila de Marcus Gunn). Al iluminar la pupila del ojo afectado, hay menos reacción a la luz e incluso puede producirse un reflejo paradójico con midriasis. Este sería el único dato clínico que podríamos obtener si el paciente estuviera adormecido, ya que se trata en múltiples ocasiones de pacientes con un traumatismo grave (incluso en estado de coma), en los cuales la valoración es difícil.
El fondo de ojo es normal inicialmente, aunque al mes del traumatismo aparecerá una atrofia óptica.
La avulsión del nervio óptico consiste en la sección parcial o completa del nervio en la salida del globo ocular (a nivel de la lámina cribosa). El paciente presenta una amaurosis. En el fondo del ojo, la papila queda sustituida por una hemorragia o vacío. No existe tratamiento.
Hemorragia orbitaria y síndrome compartimental
La órbita debido a sus características anatómicas y su pequeño tamaño es propensa a que se produzcan síndromes compartimentales.
Es un espacio cerrado limitado por las cuatro paredes óseas, el globo ocular y el tabique orbitario. Un volumen pequeño de hemorragia puede producirlo.
La disminución de la visión, con dolor, diplopía y proptosis son signos y síntomas de hemorragia orbitaria. Si se acompañan de un defecto pupilar aferente, pueden justificar la presencia de un síndrome compartimental orbitario agudo, que comprima el nervio óptico causando su lesión.
Si el nervio óptico o la circulación retiniana están comprometidos es necesario un tratamiento urgente para aumentar el volumen de la órbita y permitir que el globo ocular se desplace en sentido anterior. La mejoría de la agudeza visual y desaparición del efecto pupilar aferente demuestra el éxito de la intervención.
Traumatismo ocular abierto
El traumatismo ocular abierto es una grave complicación secundaria a un traumatismo. Una erosión en la piel del párpado o una hemorragia subconjuntival pueden ser la única manifestación superficial de la perforación de la esclerótica por unas tijeras o por un dardo.
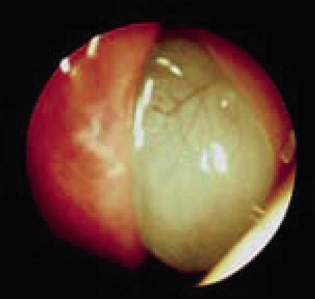
Llamamos penetración ocular (Fig. 12) a la herida realizada por un objeto punzante cuya puerta de entrada es la misma que la de salida. Llamamos perforación ocular a la herida realizada por un objeto punzante cuya puerta de entrada es diferente a la puerta de salida.
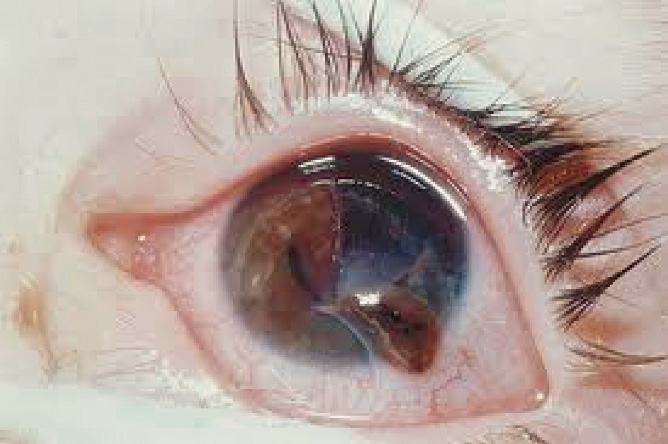
Figura 12. Penetración ocular.
Lesiones por cristales, objetos afilados (tijeras, agujas y cuchillos), lápices, perdigones, objetos lanzados a distancia y hojas de plantas o ramas de árboles obligan a exploraciones complementarias(7). La distorsión de la pupila puede ser el signo más evidente de una pequeña penetración corneal.
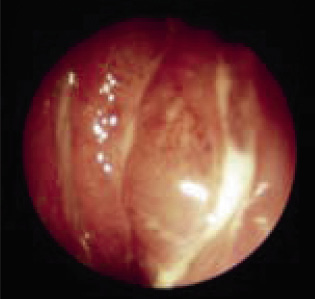
El cuerpo extraño intraocular (Fig. 13) es poco frecuente, pero hay que descartarlo mediante una radiografía simple de cráneo cuando es metálico. Si no, hay que realizar una TAC.
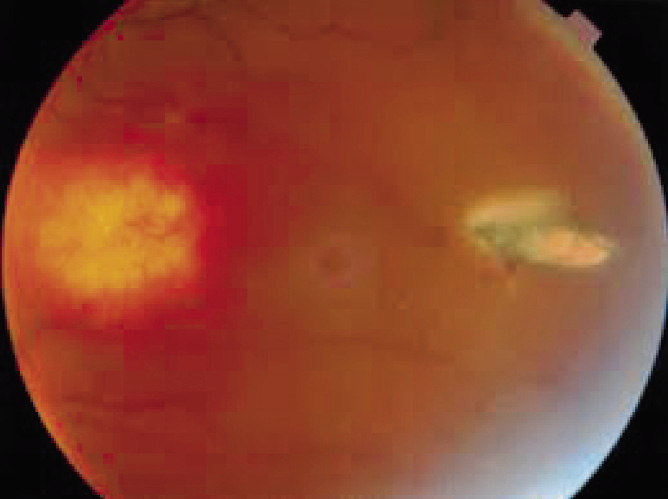
Figura 13. Cuerpo extraño intraocular.
La ruptura o estallido del globo ocular es una herida del grosor total de la pared del globo ocular provocada por un objeto romo. La laceración del globo ocular es una herida del grosor total de la pared del globo ocular provocada por un objeto afilado.
Las laceraciones tienen bordes más definidos y causan menos trastornos intraoculares. Las rupturas tienen bordes desiguales y crean una desestructuración intraocular notable.
El lugar de ruptura se suele localizar en el punto más débil de la esclera, que suele ser la zona de detrás de las inserciones de los músculos rectos.
La dificultad de explorar a niños pequeños aumenta cuando hay lesiones oculares graves. Con ayuda de los padres, se puede explorar con lámpara de hendidura o, si el trauma es abierto, con anestesia, para diagnosticar y tratar la lesión de la pared del globo ocular de grosor total. Esta exploración la debe realizar un oftalmólogo. Es muy importante que, ante una sospecha de estallido o laceración ocular, se remita al paciente a un especialista para realizar la sutura lo antes posible. Una demora de 24 horas puede suponer un empeoramiento importante del pronóstico visual.
Los síntomas son: dolor y disminución de la agudeza visual, que representa un signo de gravedad si es importante.
Los signos clínicos que se presentan ante un traumatismo ocular abierto son: hipotonía ocular, herniación de estructuras del ojo por la herida corneal o escleral (tejido intraocular prolapsado como: cristalino, iris, cuerpo ciliar, retina, coroides y humor vítreo).
Existe riesgo de endoftalmitis y también de oftalmía simpática (respuesta inmunológica del organismo al contacto con tejido coroideo expuesto en el momento de la lesión). Suele aparecer en el ojo sano tras años del traumatismo, no se puede prevenir y puede causar amaurosis por una panuveítis extensa. El riesgo de estas temibles complicaciones aumenta de forma exponencial a medida que se demora el tratamiento del traumatismo ocular abierto. La enucleación del ojo traumatizado evita la reacción simpática del ojo sano. Sin embargo, no la impide si se ha iniciado.
Ante la sospecha de traumatismo ocular abierto no hay que manipular el globo ocular. Hay que cerrar el ojo de manera no compresiva y derivar el paciente al oftalmólogo, que realizará bajo anestesia general la sutura.
El pronóstico visual es sumamente reservado en los traumatismos oculares abiertos.
La prevención es el mejor tratamiento de todas las lesiones oculares. En el traumatismo ocular abierto hay dolor y disminución de la agudeza visual, y signos clínicos como: hipotonía ocular, herniación de estructuras del ojo a través de la herida, riesgo de endoftalmitis y de oftalmía simpática.
Niño maltratado
No siempre podemos apreciar las manifestaciones oculares del niño maltratado. Estas se pueden encontrar hasta en un 35-45% de los casos(11-13).
La lesión por sacudida o niño zarandeado (shaken baby síndrome) constituye una de las manifestaciones más importantes de los malos tratos infantiles. Son pacientes menores de 3 años y, sobre todo, de 12 meses. Se detectan lesiones intracraneales y oculares. Como lesión intracraneal, aparece hematoma subdural y hemorragia subaracnoidea, y las manifestaciones oculares se caracterizan por hemorragias retinianas (como manifestación más frecuente) en la región macular, pudiendo ser extensas y ocupar todo el fondo de ojo, y preretinianas, sin signos o con mínimos signos de traumatismos externos. La cabeza del lactante, relativamente grande en relación al cuerpo, y la inestabilidad de la musculatura del cuello, favorecen esta patología(8).
También pueden detectarse: equimosis periorbitarias, cataratas, subluxación del cristalino, hemorragias vítreas y coroideas.
Ocurren hemorragias retinianas secundarias a traumatismos durante el parto, que son comunes en los recién nacidos, pero no persisten tras el primer mes de vida.
Las hemorragias retinianas detectadas, superado el periodo posparto, son diagnósticas de maltrato. Raramente se pueden observar por mecanismos distintos al de la sacudida, aunque no debemos olvidar los trastornos de coagulación. Las hemorragias intraoculares suelen ser bilaterales (20% unilaterales). Pueden debutar con: convulsiones, letargia, vómitos, fallo de medro y coma.
El 35% de estos pacientes sufre posteriormente ceguera o deterioro visual.
La sospecha de maltrato infantil obliga al pediatra u oftalmólogo a efectuar la denuncia correspondiente.
Las hemorragias retinianas en el niño son sugestivas de maltrato.
Traumatismos oculares deportivos
Los traumatismos oculares deportivos son más frecuentes en niños y adolescentes.
Los deportes que los pueden producir son: las artes marciales, los deportes con raquetas o aquellos en los cuales intervienen pelotas en movimiento rápido, tiro con arco y deportes con contacto físico como son el fútbol y el baloncesto, así como el boxeo y la lucha. No se puede olvidar el paint-ball (juego de guerra con balas de pintura).
Hay protectores oculares para todos los deportes. El material de elección para las gafas protectoras es el policarbonato. Es resistente al impacto con excelentes propiedades ópticas. Existen también combinaciones de cascos y caretas.
Otras entidades(7)
• Retinopatía de Purtscher: es una entidad caracterizada por hemorragias retinianas múltiples y disminución secundaria de la visión debido a traumatismo cefálico o aplastamiento torácico o toracoabdominal. Da lugar a un cuadro exudativo y hemorrágico en el fondo de ojo. Estos signos desaparecen con rapidez.
• Embolia grasa: se puede manifestar tras politraumatismos a nivel de pelvis y extremidades inferiores. Existe un intervalo libre y aparece una disminución de la visión.
• Retinopatía por valsalva: se producen hemorragias preretinianas o intraretinianas superficiales por el esfuerzo del vómito, la tos o incluso el levantamiento de pesas por adolescentes. Sin embargo, no ocurren hemorragias retinianas debido a los vómitos en los lactantes o niños pequeños. El pronóstico es excelente con recuperación completa.
Bibliografía
Los asteriscos reflejan el interés del artículo a juicio del autor.
1.*** Banta James T. Traumatismos oculares. Elsevier España; 2008.
2.*** Bargueño C, Colunga M, González E, Cienfuegos S, Díez-Lage A, Diab M. Traumatismo oculares en edad pediátrica. An Esp Pediatr. 1998; 48: 625-30.
3.** Scott E, Olitsky D, Hug Laura S, Plummer Erin D, Stahl Michelle M, Ariss Timothy P, Lindquist and Smith L. Disorders of the Eye. Chapter 618-35: 2569-615. En: Nelson Textbook of Pediatrics. Kliegman, Stanton, ST Geme, Schor. 20th edition Saunders. Elsevier; 2015. Part XXVIII.
4.** Protective eyewear for Young athletes. Ophtalmology. 2004; 111: 600-3.
5.*** Catalano RA. Traumatismos oculares y su prevención. En: Clínicas pediátricas de Norteamérica. Oftalmología. Ed. Interamericana; 1993; 4: 915-30.
6.*** Martin N, Gil-Gibernau JJ. Traumatismes oculars: diagnostic i tractament. Pediatria Catalana. 2002; 62: 298-305.
7.*** Gil-Gibernau JJ. Traumatismos oculares y de los anexos. Capítulo XII. En: Tratado de Oftalmología Pediátrica. Gil Gibernau JJ. Barcelona: ed. Scriba; 1997. p. 247-59.
8.*** Traumatismo ocular en la infancia. Oftalmología pediátrica y estrabismo. American Academy of Ophtalmology. 2008. Capítulo 30. p. 441-9.
9.*** Khaw PT, Shan P, Elkington AR. ABC of eyes. Injury to the eye. BMJ. 2004; 328: 36-8.
10.** Kanski JJ. Oftalmología clínica. Traumatismos. Capítulo 19. 5ª ed. Editorial Elsevier; 2009. p. 675-96.
11.** Miller K, Apt L. The eyes. Eye trauma and emergencies. En: Rudolph’s pediatrics. The McGraw-Hill; 2003. 26: 2412-16.
12.*** Placeres J, Mobayed J, Mengual E. Traumatismos oculares en edad pediátrica. En: Actualizaciones en oftalmología pediátrica. Esteve. 2003; 2: 115-26.
13. Casanovas Gordó JM, Martín Gómez V. Traumatismos oculares. Pediatr Integral, 2013; XVII(7): 507-19.
Bibliografía recomendada
- Banta James T. Traumatismos oculares. Elsevier España; 2008.
Tratado que describe de forma exquisita los traumatismos oculares. Acompañado de un material iconográfico excelente.
- Martín N, Gil-Gibernau JJ. Traumatismes oculars: diagnostic i tractament. Pediatria Catalana. 2002; 62: 298-305.
Excelente artículo publicado en Pediatria Catalana en el año 2002, en el cual se hace un repaso a los traumatismos oculares en una sección de la revista dedicada a formación continuada para pediatras generales.
- Gil-Gibernau JJ. Traumatismos oculares y de los anexos. Capítulo XII. En Tratado de Oftalmología Pediátrica. Gil Gibernau JJ. Barcelona: ed. Scriba; 1997. p. 247-59.
Tratado de oftalmología, referente de la oftalmología pediátrica, con un capítulo dedicado a los traumatismos oculares en la infancia.
- Khaw PT, Shan P, Elkington AR. ABC of eyes. Injury to the eye. BMJ. 2004; 328: 36-8.
Este artículo publicado en BMJ, tiene un gran interés, porque además de aportar un resumen de los traumatismos oculares, se añaden unas imágenes, muy sugerentes de toda la patología traumática ocular.
| Caso clínico |
|
Varón de 10 años de edad que acude de urgencias por presentar: dolor ocular, lagrimeo, fotofobia intensa y visión borrosa en ambos ojos, de inicio brusco esta mañana al despertarse. Como antecedente de interés, estuvo esquiando con sus padres el día anterior. A la exploración ocular, presenta hiperemia ciliar intensa y una epiteliopatía punteada superficial moderada-grave en ambos ojos. Diagnóstico: Queratitis fotoeléctrica. Tratamiento: Oclusión primeras 24 horas. Eritromicina pomada cada 8 horas. Ciclopléjico colirio cada 8 horas.
|