|
| Temas de FC |
S. Macías Franco, P. Rozas Reyes
Licenciadas en Medicina. Especialistas de Área en Oftalmología. Hospital Universitario Central de Asturias
| Resumen
Los problemas congénitos son el resultado de alteraciones que interrumpen el desarrollo intrauterino, secundarios a condiciones genéticas, infecciosas, sustancias químicas o radiación. La formación del ojo y sus anejos comienza en la 4ª semana de gestación y la diferenciación de las estructuras del ojo no culmina hasta después del primer año de vida; por lo que, dependiendo del momento en que se produzcan las anomalías, estas podrán afectar a cualquier nivel del sistema visual, desde los párpados hasta el propio nervio óptico. Se realiza una revisión de la patología congénita oftalmológica, que incluye: las malformaciones por alteraciones en el desarrollo embrionario e infecciones adquiridas durante el periodo fetal, con especial énfasis en la exploración que el pediatra debe realizar en el recién nacido. |
| Abstract
Ocular congenital problems are the result of disruptions of the intrauterine development caused by genetical conditions, infections, chemical substances or radiation. Eye formation begins at fourth week of gestational age and structures differentiation will not be over until the first year of age. Depending on the moment these alterations occur any structure can be affected, from the eyelids to the optic nerve. We perform a review of the ophthalmic congenital disorders that include malformations due to embryological development alterations as well as acquired infection during the fetal period. We also focus on the exploration that the pediatrician must carry out in the new born in order to diagnose these entities. |
Palabras clave: Alteraciones oculares congénitas; Malformaciones oculares; Cataratas; Infecciones congénitas
Key words: Congenital eye disorders; Ocular malformations; Cataracts; Congenital infections
Pediatr Integral 2018; XXII (1): 6 –15
Patología congénita ocular
Introducción
El término congénito, del latín Cum (con) genitus (engendrado), se refiere a una condición con la que un individuo nace.
Es importante conocer el desarrollo embriológico de la órbita, ojo y sus anejos, para comprender gran parte de la patología que vamos a tratar a continuación. El espectro es muy amplio, tanto por la diversidad de estructuras que pueden estar implicadas como por la gravedad de la afectación que es muy variable. Esto es debido al solapamiento anatómico y funcional, embriológico y genético, del ojo y sus anejos, que hacen de la estandarización o clasificación de las enfermedades oftalmológicas congénitas una tarea difícil; por lo que, explicarlas como entidades aisladas, podría resultar confuso a la par que abarcaría mucho más de lo que concierne al objetivo de esta revisión, que pretende guiar al pediatra en su práctica clínica.
Los tratamientos irán enfocados a mejorar la calidad de vida del paciente. El desarrollo visual binocular normal será un reto en muchas ocasiones, una carrera a contrarreloj, donde el tiempo y el crecimiento del niño nos obligará a diagnosticar y a actuar desde tempranas edades.
Embriología (Organogénesis) del ojo y sus anejos (Tabla I)

El desarrollo del sistema visual comienza en la 4ª semana de gestación. Las estructuras oculares procederán del ectodermo y mesodermo sin participación del endodermo.
El primer esbozo del sistema ocular se aprecia a partir de la 3ª-4ª semana de gestación con la aparición de las vesículas ópticas y tallos ópticos a partir del prosencéfalo o cerebro primitivo (neuroectodermo). Dichas vesículas, al entrar en contacto con el ectodermo superficial, inducen la formación de las vesículas o cúpulas cristalinianas hacia la 4ª- 5ª semana, de donde procederán el cristalino y el epitelio corneal. A su vez, las vesículas ópticas se invaginan adquiriendo forma de cáliz y englobando a las cristalinianas. Otra invaginación también se produce en la zona inferior del ojo, por lo que originalmente no será una estructura cerrada.
En este punto vemos un “ojo primitivo” con una zona más anterior, donde el desarrollo del ectodermo superficial y las células de la cresta neural dará lugar a la diferenciación de las estructuras más anteriores del ojo (párpados, córnea, iris, malla trabecular, cuerpo ciliar, etc.) y sistema lagrimal; y otra posterior, de procedencia neuroectodérmica, configurada por la cavidad vítrea, una retina de doble capa y el nervio óptico. La apertura inferior (fisura coroidea) permite la invasión del mesénquima, derivado mesodérmico y origen de las estructuras vasculares del ojo, los vasos hialoideos que involucionarán hasta desaparecer durante la vida fetal y los vasos definitivos que permanecerán tras la regresión de los primeros.
El cierre de la fisura coroidea acontece sobre la 7ª-8ª semana, aunque un pequeño creciente inferior permanece sin cerrar hasta estadios más avanzados(1).
Malformaciones “globales” del ojo
La interrupción del desarrollo precoz afecta a diferentes estructuras oculares concomitantemente con consecuencias devastadoras(2).
• Ojo de tamaño pequeño: a diferencia del nanoftalmo que se trata de un ojo de pequeñas proporciones, pero con normal funcionamiento, el ojo microftálmico (Fig. 1) será patológico.
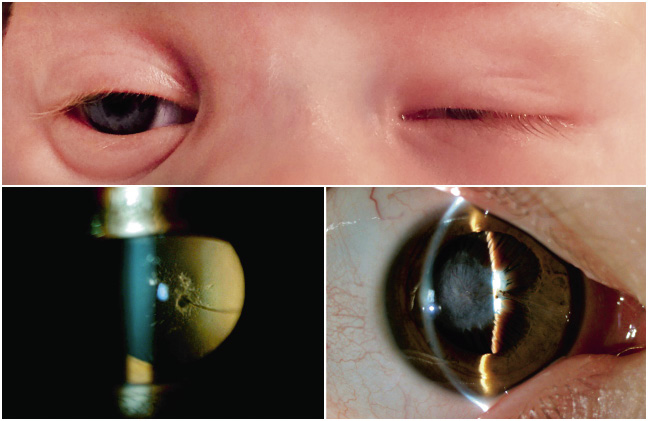
Figura 1. Anomalías globales. Superior: microftalmos izquierdo. Inferiores: persistencia de vasculatura fetal leve con mancha de Mittendorf y cordón fibrovascular (izquierda) y severa con catarata completa y elongación de procesos ciliares (derecha).
Asocia desde opacidades corneales hasta defectos retinianos o del nervio óptico, dependiendo del momento de “interrupción del desarrollo”. Si este se detiene antes de la 4ª semana, no se forma la vesícula cristaliniana y encontraremos un “esbozo de ojo” o anoftalmos, la forma más severa. En cambio, a partir de la 8ª semana o inicio del periodo fetal, las anomalías serán menores y compatibles con buenas visiones.
• Persistencia de vasculatura fetal: incompleta regresión del sistema vascular fetal(3). La mayoría aparece como formas leves, donde se evidencian vestigios de la arteria hialoidea como en la “mancha de Mittendorf”, pequeña opacidad blanca sobre la cápsula posterior del cristalino frecuentemente asintomática, o manifestarse con persistencia del vítreo primario, microftalmos, catarata y/o desprendimiento de retina, que suponen mal pronóstico (Fig. 1).
• Colobomas: cierre incompleto de estructuras. Los colobomas palpebrales se tratan de “muescas”, frecuentemente en párpado superior, aunque también aparecen en el párpado inferior, como es característico en el síndrome de Treacher Collins (Fig. 2).

Figura 2. Anomalías palpebrales. Superiores: epibléfaron (izquierda), coloboma inferior en síndrome de Treacher Collins (derecha). Centrales: epicanto y ptosis derecha antes (izquierda) y a la semana de la cirugía de la ptosis (derecha). Inferiores: hemangioma profundo antes (izquierda) y tras tratamiento con propanolol oral (derecha).
Por otro lado, los colobomas que afectan al globo ocular se deben a un defecto en la fusión de los extremos de la fisura embrionaria. En los colobomas de iris, la pupila no se ve redondeada, sino desviada inferiormente por una falta del mismo (Fig. 3 y 6).
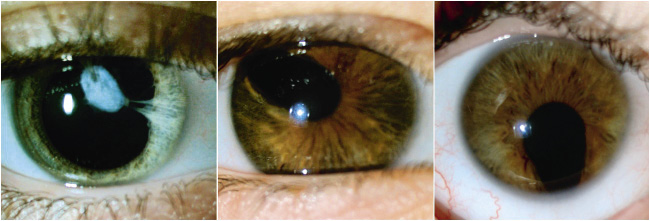
Figura 3. Alteraciones en el iris (de izquierda a derecha): sinequia y catarata, corectopia en una disgenesia del segmento anterior, coloboma de iris.

Figura 6. Superior: leucocoria en ojo microftálmico y coloboma. Inferior: retinoblastoma.
Es necesaria la exploración de fondo de ojo para descartar la afectación coriorretiniana y del nervio, de amplitud variable y que marcará el pronóstico visual. Pese a que la mayoría son esporádicos, es importante el despistaje de otras anomalías oculares y síndromes sistémicos asociados, donde el más frecuente es el síndrome CHARGE (Coloboma, alteraciones cardiacas H, Atresia de coanas, anomalías Genitourinarias y auditivas E).
• Aniridia: se diagnostica por la ausencia o falta del iris. Es debido a mutaciones genéticas en el gen PAX6 (descritas >130 mutaciones) o en el WT1 (deleción), localizados ambos en el cromosoma 11. De carácter autosómico dominante en 2/3 de los casos, con penetrancia completa y expresividad variable, afecta a 1/60.000-100.000 RN vivos. No hay que pensar en la aniridia como una simple falta de iris, ya que asocia: catarata, hipoplasia del nervio óptico y macular y nistagmo por baja visión. Las alteraciones corneales y el glaucoma son de difícil manejo y suelen aparecer en la adolescencia-segunda década. La importancia de su diagnóstico radica en su asociación con el tumor de Wilms y/o síndrome WAGR (tumor de Wilms, aniridia, anomalías genitourinarias y retraso mental).
• Criptoftalmos: “Ojo enterrado” y disfuncional con queratinización corneal. Defecto en la formación palpebral, lo encontramos en el síndrome de Fraser (criptoftalmos, sindactilia, genitales ambiguos, labio leporino, fisura palatina, hernias y malformaciones sistémicas).
• Ciclopía: ojo único y central por indivisión del prosencéfalo. Extremadamente infrecuente por incompatibilidad con la vida de muchas de las malformaciones concomitantes.
Anomalías congénitas de párpados y vías lagrimales(4)
Grueso importante donde podemos ver desde defectos puramente estéticos hasta incapacitantes. Las anomalías más frecuentes son: el epicanto, epibléfaron y ptosis (Fig. 2).
• Causas palpebrales de pseudoestrabismo: el epicanto se trata de la existencia de un pliegue cutáneo entre párpado superior e inferior frecuente en raza asiática y síndrome de Down o Turner. Simula, en muchos casos, un estrabismo convergente, por lo que muchas veces son derivados a la consulta. Desaparece o disminuye con el crecimiento. Lo vemos asociado al aumento de la distancia intercantal (telecanto).
• Anomalías del borde palpebral: el epibléfaron se define como un pliegue redundante de piel en párpado inferior que rota las pestañas y hacen que estas rocen con el globo ocular. Casi siempre es asintomático y desaparece con el desarrollo facial a diferencia del entropión, condición en la cual existe rotación interna del borde libre palpebral, sintomático y puede precisar cirugía. En el ectropión, el párpado estará rotado hacia afuera, lo que causa exposición conjuntival y falta de cobertura ocular. El euribléfaron combina el acortamiento de piel con una laxitud horizontal, el resultado es el de un ectopión de la zona más temporal.
• Ptosis: párpado “caído” por desarrollo incompleto del músculo elevador del mismo, como consecuencia de un parto traumático o en el contexto de una parálisis del III par. Cuando la oclusión del ojo afecta al eje visual, existe riesgo de ambliopía por deprivación, por lo que el tratamiento con parches desde el nacimiento puede ser necesario y la valoración quirúrgica precoz.
• Hemangioma: tumoración vascular que rara vez se presenta al nacimiento. Se ve como una lesión plana y roja (superficiales) o elevada y violácea (profundos). Sus complicaciones incluyen: ptosis, limitaciones en los movimientos oculares o proptosis si son muy grandes, por lo que serán remitidos para valorar tratamiento con propanolol oral o tópico(5) (Fig. 2).
• Obstrucción congénita del conducto nasolagrimal (OCNL): causa más frecuente de epífora en el recién nacido y lactante por incompleta canalización del sistema de drenaje lagrimal, frecuentemente a nivel distal. Además de lagrimeo desde el nacimiento, se manifiesta con conjuntivitis de repetición. Se recomienda la observación y tratamientos con masajes y lavados de las secreciones, así como antibioticoterapia tópica en los episodios infecciosos hasta el primer año de vida, ya que se resuelven en un 90%. De lo contrario, deben ser valorados por el oftalmólogo para la realización de sondaje, dilatación con balón, intubación o, en casos más graves y sin resolución, dacrioplastia o dacriocistorrinostomía.
Anomalías congénitas del segmento anterior
Alteraciones en el desarrollo fetal causarán disgenesias (desdiferenciaciones) con afectación desde la córnea al cristalino(6).
• Anomalías del iris (Fig. 3): manifestaciones iridianas como: la desviación de la pupila (corectopia), sinequias a córnea o cristalino y múltiples pupilas (policoria), son raras de manera aislada y suelen presentarse como parte de las manifestaciones de algunas disgenesias del segmento anterior. La membrana pupilar persistente se refiere a una banda de iris que atraviesa la pupila y puede estar adherida a la cápsula anterior del cristalino.
• Anomalías corneales (Fig. 4):

Figura 4. Córnea y cataratas. Superiores: anomalía de Peters (izquierda), quiste dermoide (derecha). Inferiores: distintas manifestaciones de cataratas congénitas. Vemos la heterogeneidad en la presentación de la opacidad cristaliniana.
- Quistes dermoides: son coristomas, derivados ectodérmicos de aspecto rosado y brillante que pueden contener pelo y otras estructuras ectodérmicas. Se localizan en el limbo corneoescleral y su extirpación temprana depende de las molestias o complicaciones debidas a un tamaño considerable.
- Anomalía de Peters: opacidad corneal uni o bilateral, con o sin unión (sinequias) al iris o incluso al cristalino. Se trata de una disgenesia de las capas posteriores de la córnea y su separación del cristalino. El trasplante corneal se considera el tratamiento de elección, donde las tasas de supervivencia del injerto y recuperación visual son muy variables(7).
- Otras alteraciones corneales congénitas: las úlceras por trauma durante el parto cursan con ojo rojo, epifora, fotofobia e irritabilidad. Las distrofias corneales son infrecuentes y no suelen detectarse al nacimiento.
• Anomalías del cristalino (Fig. 4):
- Catarata: patología congénita más frecuente del cristalino y considerada la causa más frecuente de ceguera tratable. Se define como una opacidad del cristalino que puede ser parcial o completa y que interferirá variablemente en la visión. Su uni o bilateralidad es un dato importante a considerar a la hora de buscar la causa responsable (Tabla II), así en las bilaterales, encontramos frecuentemente un patrón hereditario autosómico dominante, por lo que una buena anamnesis y exploración de los progenitores puede evitar la realización de pruebas complementarias innecesarias (Algoritmo 1).

También aparecen, aunque en menor medida, en el contexto de enfermedades genéticas, metabólicas, teratógenos e infecciones maternas durante el embarazo. Por otro lado, las cataratas unilaterales nos hacen pensar más en malformaciones oculares. Aun existiendo un gran número de factores etiopatogénicos, es importante destacar que la gran mayoría se consideran idiopáticas, un 60% en bilaterales y hasta un 80% en unilaterales. En estas últimas, se encuentran con relativa frecuencia vestigios de la arteria hialoidea, indicio de que posiblemente la persistencia de la vasculatura fetal leve sería el origen de muchas de las cataratas catalogadas como idiopáticas. Lo mismo ocurre con las bilaterales, se cree que muchas pueden cursar con alteraciones genéticas, por lo que su etiología puede estar infradiagnosticada(8).
• Otras anomalías congénitas del cristalino: la ausencia de cristalino congénita o afaquia es una condición infrecuente. La ectopia o subluxación se deberá a un defecto de la zónula de Zinn (ligamento suspensorio del cristalino) que causa desplazamientos del mismo, típico de la enfermedad de Marfan y de la homocistinuria. Microesferofaquia hará referencia a un cristalino pequeño, esférico, ectópico y relacionado con la enfermedad de Weill-Marchesani. La existencia de una protrusión de apariencia cónica con o sin opacidad y característica de la enfermedad de Alport se conoce como lenticono.
• Glaucoma(9) (Fig. 5):

Figura 5. Glaucoma congénito. Superior: buftalmos izquierdo por glaucoma congénito. Inferior: gonioscopia.
- Glaucoma congénito primario: es detectado desde el nacimiento-primeras semanas de vida y no está asociado a otras anomalías oculares, sistémicas o síndromes. Representa más de la mitad de los glaucomas pediátricos. La mayoría son esporádicos y una pequeña parte de herencia recesiva, por lo que aumenta su incidencia en regiones con alta tasa de consanguinidad. Se trata de una iridotrabeculodisgenesia, donde la afectación de la malla trabecular, responsable del drenaje del humor acuoso, provoca un aumento de resistencia a la salida del mismo, con elevación de la presión intraocular y daño del nervio óptico. Cuando aparece entre el primer y tercer año de vida, hablaremos de glaucoma infantil, aunque se trate de la misma etiopatogenia. La tríada clásica del glaucoma congénito se define por: epífora, fotofobia y blefarospasmo, asociado o no a pérdida de transparencia corneal, que puede verse desde “turbia” a un color azulado característico. El término buftalmos (“ojo de buey”) se utiliza para describir el aumento del tamaño de la córnea y globo ocular, indicios de enfermedad avanzada. Ante sospecha de glaucoma, se debe realizar lo antes posible una exploración oftalmológica bajo anestesia. El tratamiento de elección es la cirugía.
- Otros glaucomas primarios congénitos: glaucomas primarios asociados a otras anomalías oculares como en la aniridia o en la anomalía de Peters, y/o sistémicas como en el síndrome de Axenfelt-Rieger (anomalías dentales, pliegue umbilical) o enfermedad de Sturge-Weber (displasia venosa facial con “mancha de vino de Oporto”). Su manejo difiere respecto al del glaucoma congénito primario, por lo que es importante tenerlos en cuenta.
- Glaucomas secundarios: no se producen por una disgenesia per se, sino debidos a alteraciones a otros niveles del ojo que pueden comprometer al drenaje del humor acuoso y que habitualmente no se manifiestan al nacimiento.
Anomalías congénitas del segmento posterior
Sospecha en prematuros, infecciones durante el embarazo, otras anomalías oculares o exploración con fulgor pupilar anómalo, estrabismos o nistagmos.
• Retinopatía del prematuro (ROP): es una de las principales causas de ceguera evitable. La vascularización retiniana se completa hacia la semana 36-40. En ella están implicados factores angiogénicos, como el IGF y el VEGF, que se descontrolan en la ROP, produciendo una vascularización anómala y patológica. Su incidencia es inversamente proporcional a edad gestacional y peso al nacimiento (81,6% de niños <1.000 g y 46,9% de niños > 1.000 g). El aumento de la supervivencia, gracias a la mejoría en los cuidados y técnicas, ha provocado un aumento de la prevalencia de la ROP. Otro factor de riesgo es la exposición a O2. Los protocolos de screening consensuados entre pediatras y oftalmólogos han ayudado a su detección precoz(10). Dependiendo del estadio de la enfermedad, la actuación irá desde la observación hasta el tratamiento con antiangiogénicos intravítreos(11), láser o cirugía en casos más graves.
• Retinoblastoma: su incidencia de 1/15.000 RN lo convierte en el tumor ocular maligno más frecuente en niños y el tercer tumor ocular maligno más frecuente tras el melanoma y las metástasis. El 90% se manifiesta antes de los 3 años y puede estar presente al nacimiento. 2/3 de los casos son unilaterales y 1/3 bilaterales. El tumor resulta de dos mutaciones en el gen RB1 (cromosoma 13), que codifica una proteína reguladora de la proliferación celular, por lo que habrá un crecimiento descontrolado. El 40% se consideran hereditarios, la primera mutación se hereda de uno de los padres y la segunda se produce durante el desarrollo embrionario. El signo más frecuente es la leucocoria (Fig. 6), seguido del estrabismo, pero puede simular una celulitis preseptal, manifestarse como glaucoma doloroso o cambios en el color del iris. La tasa de supervivencia en nuestro medio es >90%, donde en 2/3 el ojo se conservará, mientras que en países en vías de desarrollo la supervivencia es tan solo del 45%. El tratamiento de elección es la quimioterapia intraarterial en la arteria oftálmica(12). Otros tratamientos son: la quimioterapia sistémica e intravítrea, braquiterapia, crioterapia, termoterapia y la enucleación, que consiste en la extracción del globo ocular. Dependerán del estadio de la enfermedad y del estado del paciente.
• Otras alteraciones en el segmento posterior: la exploración del fondo de ojo no es rutinaria en el recién nacido y, muchas veces, las alteraciones se encuentran a raíz de bajas visiones en el crecimiento en la consulta del oftalmólogo. Un indicio de alteración retiniana serán los nistagmos en un ojo aparentemente normal. Así, podemos encontrar una amplia diversidad de alteraciones en el fondo de ojo como: albinismo ocular, cicatrices retinianas, desprendimientos de retina o anomalías del nervio óptico como: hipoplasias, fosetas, anomalía de Morning Glory, o colobomas.
Alteraciones congénitas de los movimientos oculares y estrabismo
Cualquier desviación ocular al nacimiento deberá ser remitida al oftalmólogo, ya que puede ser la primera manifestación de retinoblastoma.
• Estrabismo: pérdida de paralelismo en los ojos. El estrabismo en el neonato es muy infrecuente. La endotropia congénita suele manifestarse a partir de los 6 meses de vida, por lo que muchos autores no coinciden con el término de “congénito”. Sí se pueden considerar como tal los estrabismos debidos a disgenesias o desórdenes inervacionales, como el síndrome de Möbius, que cursa con parálisis concomitante del VI y VII par, o el síndrome de Duane, donde existe una inervación anómala de los músculos rectos horizontales(13).
• Nistagmo: movimientos rápidos oculares involuntarios. La manifestación al nacimiento es infrecuente. Se remitirán al oftalmólogo para buscar causas oculares o neurológicas de nistagmo.
Infecciones congénitas con afectación ocular
Dependiendo del patógeno y momento de infección de la gestante, las manifestaciones en el neonato serán variables.
Una vez familiarizados con las patologías de mayor interés para el pediatra, consideramos de importancia realizar un breve resumen de las infecciones congénitas clásicas con repercusión ocular adquiridas en el periodo fetal, por lo que este bloque se sale del esquema que hemos visto hasta ahora, ya que no se centra en la estructura ocular sino en el agente teratógeno, en este caso, los distintos microorganismos. En la tabla III describimos las infecciones congénitas clásicamente más relevantes y que cursan con afectación ocular(14).

Para finalizar, merece mención la conjuntivitis neonatorum. Esta, realmente, se trata de una enfermedad adquirida en el periodo peri y postnatal, por lo que no vamos a desarrollarla en esta revisión. Son múltiples los patógenos implicados, donde el gonococo y la clamidia serán responsables de formas muy agresivas. La profilaxis con povidona yodada o eritromicina tópica han disminuido drásticamente su incidencia(15).
Función del pediatra de Atención Primaria en la exploración en el recién nacido, detección de defectos oculares (Algoritmo 2)
Una exploración precoz minuciosa y protocolizada es fundamental en la detección de la patología oftalmológica(16).
• Epífora, secreciones: la OCNL se detecta por la presencia de secreciones en torno al borde palpebral y conjuntivitis de repetición. No debemos confiarnos, ya que también puede ser un signo de erosiones corneales o glaucoma congénito, a este suele acompañarle una intensa fotofobia y blefarospasmo. Ante la sospecha de OCNL, se podrán tratar con lavados, masajes y pomadas oftálmicas, como eritromicina, esperando su resolución. La no respuesta al tratamiento será motivo de derivación al especialista en torno al año de edad.
• Tamaño: un ojo pequeño nos alertará de un desarrollo incompleto asociado a catarata, en muchas ocasiones. Córneas grandes, nos harán sospechar de glaucoma. Las medidas de esta última no se realizan de forma rutinaria en el recién nacido, además de la dificultad que ello implica, incluso para el oftalmólogo pediátrico, que muchas veces precisará de la exploración bajo anestesia para lograrlo. Es muy útil la comparación con el ojo contralateral, ya que las patologías relacionadas suelen ser unilaterales, por lo que la simetría puede ser indicador de normalidad, aunque no descarta la enfermedad y podemos encontrar glaucomas congénitos y ojos microftálmicos bilaterales.
• Alteraciones palpebrales: llamativas y fáciles de detectar. Nos fijaremos en la posición del borde palpebral respecto al globo ocular, continuidad del mismo y la orientación de las pestañas y roce de estas con el ojo. Una ptosis congénita o un defecto donde el eje visual pueda verse comprometido, la derivación preferente al oftalmólogo está más que justificada, ya que su corrección precoz y el inicio de oclusiones con parche o penalización evitarán la ambliopía.
• Transparencia de medios: la córnea transparente permite ver con claridad el color del iris y la pupila. Una pérdida de esa transparencia nos alertará sobre una posible disgenesia del segmento anterior. Será característico el color azulado de la misma en el glaucoma congénito, aunque no tiene por qué cumplirse, siempre dependerá de cuán avanzada esté la enfermedad.
• Valoración pupilar y motilidad intrínseca: las pupilas deben ser redondas, del mismo tamaño y isorreactivas. Su deformidad hará pensar en colobomas o disgenesias del segmento anterior. Una midriasis excesiva arreactiva es indicio de aniridia.
• Movimientos oculares: podrán valorarse llamando la atención del niño desde diferentes posiciones. Las desviaciones y limitaciones en los movimientos oculares nos harán sospechar de parálisis-paresias de los pares creaneales oculomotores.
• Test de Brückner: consiste en proyectar la luz del oftalmoscopio hacia la pupila. El reflejo del ojo deberá ser naranja e isointenso en ambos ojos. Ante opacidades, tanto corneales como cristalinianas, veremos una opacidad negra por retroiluminación de este reflejo, causa justificada para la derivación preferente al oftalmólogo. La leucocoria (Fig. 6) será la visualización de este reflejo de color blanquecino, esto será causa de derivación urgente al especialista para descartar el retinoblastoma. También, aparece en colobomas por el reflejo escleral directo debido a “falta de retina”.
• Protocolos de screening de la retinopatía de la prematuridad: debe existir una buena comunicación entre el pediatra y el oftalmólogo. Lo ideal es que sean siempre las mismas personas las que realicen los controles y la asignación de un día semanal para ello.
Bibliografía
Los asteriscos reflejan el interés del artículo a juicio del autor.
1. Moore K, Persaud T. Embriología Clínica (9ª ed.). Ed. Elsevier; 2013.
2.** Rachitskaya A, Traboulsi E. Congenital ocular malformatioms. En: Wilson ME, Saunders R, Trivedi R. Pediatric Ophthalmology. A current Thought and a practical guide. Ed. Springer. Ed. Springer, 2009. p. 287-310.
3. Shastry BS. Persistent hyperplastic primary vitreous: congenital malformation of the eye. Clin Exp Ophthalmol. 2009; 37: 884-90.
4.*** Trueba A. Patología congénita de la vía lagrimal y patología palpebral. Pediatr Integral. 2013; XVII(7): 463-76.
5.** Baselga Torres E, Bernabéu Wittel J, van Esso Arbolave D, Febrer Bosch M, Carrasco Sanz Á, de Lucas Laguna R, et al. Consenso español sobre el hemangioma infantil. Anales de Pediatría. 2016; 85: 256-65.
6. Stahl ED. Anterior Segment Dysgenesis. Int Ophthalmol Clin. 2014; 54: 95-104.
7. Rao K, Fernandes M, Gangopadhyay N, Vemuganti G, Krishnaiah S, Sangwan V. Outcome of Penetrating Keratoplasty for Peters Anomaly. Cornea. 2008; 27: 749-53.
8. Rahi JS, Dezateux C. British Congenital Cataract Interest Group. Measuring and interpreting the incidence of congenital ocular anomalies: lessons from a national study of congenital cataract in the UK. Invest Ophthalmol Vis Sci. 2001; 42: 1444-8.
9.** Abu-Amero K, Edward D. Primary Congenital Glaucoma [Internet]. Ncbi.nlm.nih.gov. 2017 [cited 15 November 2017]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1135/.
10.** Ferrer Novella C, et al. Programa de cribado para la retinopatía del prematuro en España. Arch Soc Esp Oftalmol. 2013; 88: 184-8.
11.** Sankar MJ, Sankar J, Mehta M, Bhat V, Srinivasan R. Anti-vascular endothelial growth factor (VEGF) drugs for treatment of retinopathy of prematurity. Cochrane Database Syst Rev. 2016; 2. Review.
12. Gigorovski N, Lucena E, Mattosinho C, Parareda A, Ferman S, Català J, et al. Use of intra-arterial chemotherapy for retinoblastom: results of a survey. Int J Ophthalmol. 2014; 7: 726-30.
13. Gutowsky NJ, Chilton JK. The congenital cranial dysinnervation disorders. Arch Dis Child. 2015; 100: 678-81.
14. Protocolos de infecciones de transmisión vertical y neonatales | UPIIP [Internet]. Upiip.com. 2017 [cited 15 November 2017]. Available from: http://www.upiip.com/es/docencia/protocolos-de-infecciones-de-transmisi%C3%B3n-vertical-y-neonatales.
15.** Badia J, Figaró C, Domingo M, Aldecoa V. Infecciones congénitas. Pediatr Integral. 2014; XVIII(6): 356-66.
16.*** Wright KW, et al. Pediatric Ophthalmology for Primary Care. American Academy of Pediatrics; 2008. p. 143-51, 243-52.
Bibliografía recomendada
– Rachitskaya A, Traboulsi E. Congenital ocular malformations. En: Wilson ME, Saunders R, Trivedi R. Pediatric Ophthalmology. A current Thought and a practical guide. Ed. Springer. Ed. Springer, 2009. p. 287-310.
Capítulo fácil de entender sobre malformaciones oculares congénitas.
– Trueba A. Patología congénita de la vía lagrimal y patología palpebral. Pediatr Integral. 2013; XVII(7): 463-76.
Revisión sobre patología congénita palpebral y de la vía lagrimal. Dirigido tanto a pediatras como a oftalmólogos, permite una visión sencilla y práctica del tema.
– Wright KW, et al. Pediatric Ophthalmology for Primary Care. American Academy of Pediatrics; 2008. p. 143-51, 243-52.
Excelente guía para el pediatra en su práctica clínica.
– Baselga Torres E, Bernabéu Wittel J, van Esso Arbolave D, Febrer Bosch M, Carrasco Sanz Á, de Lucas Laguna R, et al. Consenso español sobre el hemangioma infantil. Anales de Pediatría. 2016; 85: 256-65.
Consenso entre pediatras y otros especialistas, donde se incluyen recomendaciones y algoritmos de actuación en pacientes pediátricos con hemangioma infantil.
– Abu-Amero K, Edward D. Primary Congenital Glaucoma [Internet]. Ncbi.nlm.nih.gov. 2017 [cited 15 November 2017]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1135/.
Revisión de glaucoma congénito, incluida genética, con buenas referencias bibliográficas. Disponible al público online.
– Sankar MJ, Sankar J, Mehta M, Bhat V, Srinivasan R. Anti-vascular endothelial growth factor (VEGF) drugs for treatment of retinopathy of prematurity. Cochrane Database Syst Rev. 2016; 2. Review.
Revisión actualizada sobre los antiangiogénicos en la retinopatía de la prematuridad clásicamente tratada con terapia láser.
– Ferrer Novella C, et al. Programa de cribado para la retinopatía del prematuro en España. Arch Soc Esp Oftalmol. 2013; 88: 184-8.
Consenso de especialistas en retinopatía de la prematuridad, basados en evidencias y experiencia. Presenta directrices generales para realizar el cribado de la ROP, incluyendo: criterios de inclusión y exclusión, metodología de exploración y calendario de actuación.
– Sankar MJ, Sankar J, Mehta M, Bhat V, Srinivasan R. Anti-vascular endothelial growth factor (VEGF) drugs for treatment of retinopathy of prematurity. Cochrane Database Syst Rev. 2016; 2. Review.
Revisión actualizada sobre los antiangiogénicos en la retinopatía de la prematuridad, clásicamente tratada con terapia láser.
– Badia J, Figaró C, Domingo M, Aldecoa V. Infecciones congénitas. Pediatr Integral. 2014; XVIII(6): 356-66.
Revisión sobre infecciones congénitas, métodos para su detección, profilaxis y tratamientos.
| Caso clínico |
|
Niño recién nacido de 7 días de vida. Parto eutócico, edad gestacional 40 semanas y 2.200 g. Padres no consanguíneos. Madre 34 años, sin antecedentes de interés salvo leve miopía, por lo que necesita gafas para conducir. No se conocen antecedentes paternos. Remitido por pupilas muy dilatadas desde el nacimiento, movimientos oculares rápidos y sensación de que el niño “no fija bien”.
|