 |
| Temas de FC |
J. Pozo Romána,b, A. Martín Rivadac, M. Güemes Hidalgoa.
aMédico Adjunto del Servicio de Endocrinología Pediátrica del Hospital Infantil Universitario Niño Jesús de Madrid. bProfesor Asociado de Pediatría de la Universidad Autónoma de Madrid. cResidente de Pediatría y Endocrinología Pediátrica de 4º año del Hospital Infantil Universitario Niño Jesús de Madrid
| Resumen
La hipoglucemia es un trastorno metabólico frecuente en la infancia y especialmente en el periodo neonatal. Su importancia radica en que el cerebro, en condiciones normales, solo consume glucosa y no es capaz de sintetizarla ni de acumularla, por lo que las hipoglucemias pueden provocar alteraciones transitorias del funcionamiento cerebral o secuelas neurológicas definitivas. No es posible establecer un punto de corte que delimite lo que es una glucemia normal-anormal o un nivel de glucemia por debajo del cual se produzca daño neurológico. Ello se debe a que la respuesta cerebral a la hipoglucemia se produce en un rango relativamente amplio de glucemia (continuum) que puede verse alterado por la presencia de sustratos alternativos u otros factores. Las manifestaciones clínicas de la hipoglucemia varían, dependiendo, entre otros factores, de la edad del niño, pero son, en general, inespecíficas, especialmente en los recién nacidos y lactantes pequeños. |
| Abstract
Hypoglycemia is a frequent pediatric metabolic disorder, particularly common in neonates. Given that the brain ́s only fuel is glucose, and it cannot synthetize nor store it, hypoglycemia can lead to transitional or permanent brain injury. A cut-off point to discern normal from abnormal glycemia or one that causes brain damage cannot be established as brain response to hypoglycemia is produced in a wide range of glycemic values (continuum), that can be modified by alternative substrates or by diverse factors. Clinical manifestations may vary depending on aspects, such as age, but they are generally non-specific, especially in neonates and toddlers. |
Palabras clave: Hipoglucemias; Homeostasis de la glucosa; Hipoglucemia transicional; Hiperinsulinismos; Diazóxido
Key words: Hypoglycemia; Glucose homeostasis; Transitional hypoglycemia; Hyperinsulinism; Diazoxide
Pediatr Integral 2019; XXIII (2): 90.e1 – 90.e22
Hipoglucemia no diabética
Introducción
La hipoglucemia no es una enfermedad, sino la manifestación bioquímica del fracaso de alguno de los complejos mecanismos homeostáticos encargados de su mantenimiento(1). Es un trastorno metabólico frecuente en la infancia y especialmente en el periodo neonatal.
La importancia de la hipoglucemia radica en que, tanto en la vida intrauterina como a lo largo de la vida, la glucosa plasmática (GP) constituye el principal substrato energético del organismo, especialmente de los llamados órganos glucodependientes (sistema nervioso, cristalino, hematíes y médula renal), cuyo funcionamiento depende, si no exclusivamente, sí fundamentalmente del aporte continuo de glucosa.
El cerebro, aunque en situaciones especiales puede utilizar fuentes energéticas alternativas (cuerpos cetónicos, lactato), en condiciones normales solo utiliza glucosa y no es capaz de sintetizarla ni de acumularla en cantidades significativas (glucógeno astrocitario). La glucosa entra al cerebro mediante un sistema de difusión facilitada independiente de insulina, pero dependiente de la glucemia arterial; de forma que, cuando esta disminuye o existen defectos en el transportador de glucosa (GLUT), se produce glucopenia cerebral con hipoglucorraquia. En condiciones fisiológicas, la GP oscila entre 70 y 130 mg/dL y la cerebral entre 14 y 41 mg/dl, pero, cuando la GP cae por debajo de 36 mg/dl, la concentración de glucosa en el cerebro se aproxima a 0 mg/dL. Se conocen, al menos, 14 isoformas de GLUTs, con expresión diferencial en los diferentes tejidos y varios de ellos en el cerebro, de los cuales, los más importantes son: GLUT1 (barrera hematoencefálica y eritrocitos) y GLUT3 (neuronas). La mayoría de los tejidos que responden a insulina (músculo, tejido adiposo…) expresan GLUT4(2).
La hipoglucemia va a condicionar una alteración transitoria del funcionamiento cerebral, que se corrige habitualmente cuando la glucemia aumenta.
• Cuando la hipoglucemia es grave y prolongada, puede desencadenar convulsiones y, de forma excepcional, daño cerebral irreversible o incluso una arritmia cardíaca que conduzca a la muerte súbita.
• Cuando los episodios de hipoglucemia son menos graves, pero recurrentes, especialmente durante el periodo neonatal o la primera infancia, cuando el cerebro está todavía en desarrollo, pueden ocasionar secuelas neurológicas a largo plazo potencialmente graves, que oscilan desde una leve disfunción neurocognitiva a retraso mental severo, epilepsia, microcefalia, hemiparesia o afasia(3,4).
• En los niños mayores, la reiteración de hipoglucemias, habitualmente pacientes con diabetes mellitus tipo 1 (DM1) en tratamiento insulínico intensivo, puede provocar disfunciones neurocognitivas, especialmente alteraciones de la memoria, y fallo en la respuesta autonómica a la hipoglucemia, predisponiendo al desarrollo de nuevas hipoglucemias que, además, se hacen asintomáticas (hipoglucemias inadvertidas), favoreciendo su mayor gravedad y severidad(5).
Homeostasis de la glucosa
Los mecanismos homeostáticos encargados del mantenimiento de la glucemia son mecanismos de supervivencia y, por ello: múltiples, escalonados y redundantes, al objeto de que el fallo de uno o de varios de sus componentes no comprometa la vida del sujeto.
La GP deriva de tres posibles fuentes: 1) la absorción intestinal de hidratos de carbono de la dieta; 2) la liberación de los depósitos de glucosa acumulados en forma de glucógeno (glucogenolisis); y 3) la liberación de glucosa sintetizada de novo (gluconeogénesis) a partir de precursores, incluyendo: lactato, piruvato, aminoácidos (especialmente alanina y glutamina) y, en menor medida, glicerol. Muchos tejidos poseen los sistemas enzimáticos necesarios para sintetizar glucógeno e hidrolizarlo (glucógeno-sintetasa y fosforilasa), pero solo el hígado y el riñón expresan la glucosa 6-fosfatasa, la enzima necesaria para poder liberar la glucosa a la circulación. El hígado y el riñón expresan también las enzimas necesarias para la gluconeogénesis: piruvato-carboxilasa, fosfoenol-piruvato carboxiquinasa y fructosa 1-6 bifosfatasa(6).
La GP, pese a las variaciones en la actividad física y a la ingesta intermitente de nutrientes, se mantiene relativamente estable a lo largo del día; de forma que, raramente sobrepasa los 130 mg/dL postingesta, y no suele descender por debajo de los 70 mg/dL en los períodos interprandiales. La excepción la constituyen algunos niños, habitualmente menores de 4 años, que pueden presentar, al despertarse por la mañana, glucemias inferiores a 70 mg/dL y cetonemia, como consecuencia de su baja tolerancia al ayuno(7).
En condiciones normales, después de una comida (fase absortiva), las concentraciones de insulina aumentan desde 3-10 µU/mL a 20-50 µU/mL, permitiendo que el organismo consuma la glucosa procedente de los hidratos de carbono ingeridos (aumento de la utilización periférica), así como su almacenamiento en forma de glucógeno (glucogenogénesis) en diferentes tejidos, especialmente hígado y músculo. En el hígado, se favorece también la síntesis de triglicéridos, que serán finalmente transportados y almacenados en el tejido adiposo(1). La presencia de insulina en la sangre inhibe la gluconeogénesis, la lipolisis y la cetogénesis. A medida que la glucosa es consumida y almacenada (3-4 horas postingesta), la GP comienza a descender (fase postabsortiva) y los mecanismos contrarreguladores de la hipoglucemia se van poniendo en marcha de manera secuencial para prevenir el desarrollo de hipoglucemia(6,7). Estos mecanismos son mecanismos de supervivencia y, por ello, son: múltiples, escalonados y redundantes, al objeto de que el fallo de uno o de varios de sus componentes no comprometa la vida del sujeto.
1. El primer mecanismo defensivo frente a la hipoglucemia es la disminución de la secreción de insulina por las células β pancreáticas, que se inicia cuando la glucemia se aproxima a los 80-85 mg/dL. Como consecuencia, disminuye el consumo de glucosa por los tejidos periféricos insulín-sensibles, favoreciendo un adecuado aporte a los órganos glucodependientes, y se favorece la liberación de glucosa a partir del glucógeno hepático y renal (glucogenolisis). En condiciones normales, por debajo de 45-50 mg/dL de GP, la secreción de insulina estaría abolida.
2. El segundo mecanismo defensivo es la secreción de glucagón por las células alfa pancreáticas, que se pone en marcha cuando la glucosa cae justo por debajo de su nivel fisiológico (65-70 mg/dL) y que dispara la glucogenolisis hepática.
3. El tercer mecanismo de respuesta, crítico cuando la secreción de glucagón está alterada, es la secreción por la médula suprarrenal de adrenalina, que se produce, al igual que la liberación de glucagón, cuando la glucosa cae por debajo de 65-70 mg/dL. La adrenalina estimula la glucogenolisis hepática y renal, la gluconeogénesis y la lipólisis (efecto β), al tiempo que disminuye la utilización de glucosa por los tejidos periféricos insulín-sensibles (efecto β) y disminuye la propia secreción de insulina (efecto α), contrarrestando el efecto β estimulante de la secreción de insulina.
4. Un cuarto mecanismo de respuesta, menos importante en la respuesta aguda a la hipoglucemia, pero de gran importancia en el mantenimiento de la glucemia en situaciones de ayuno prolongado, es la liberación de cortisol y hormona de crecimiento (GH). Esta liberación se produce también con niveles de glucosa de 65-70 mg/dl, y ambas hormonas actúan de forma sinérgica con el glucagón y la adrenalina, incrementando los niveles de glucemia. La GH estimula la gluconeogénesis y la lipolisis; mientras que, el cortisol estimula preferentemente la gluconeogénesis.
5. Cuando la glucemia disminuye por debajo de 50-55 mg/dl, aparecen los síntomas de hipoglucemia y, entre ellos, y como consecuencia de una respuesta simpático-adrenal más intensa, la ansiedad por ingerir hidratos de carbono, una conducta defensiva que induce el aumento de los aportes exógenos de glucosa. Por último, se ha postulado que, cuando las cifras de glucemia disminuyen por debajo de 50 mg/dL, otros factores no hormonales, como estímulos neurales directos del cerebro sobre el hígado o la postulada “autorregulación hepática de la glucosa”(8), podrían actuar como mecanismos contrarreguladores complementarios.
Respuesta fisiológica al ayuno(1,2,6,9)
Los mecanismos homeostáticos de la glucosa en el niño, fuera del período neonatal, son esencialmente idénticos a los del adulto, pero los lactantes y niños pequeños, a diferencia de niños mayores y adultos, son incapaces de mantener la glucemia tras ayunos relativamente cortos (24-36 horas), debido a que: “cuanto más pequeño es el niño, mayor es su consumo proporcional de glucosa, menores sus depósitos de precursores neoglucogénicos y más vulnerable su cerebro a la hipoglucemia”.
En condiciones de ayuno (periodo postabsortivo), la interacción entre insulina y hormonas contrarreguladoras mantiene los niveles de GP relativamente estables:
• Si el ayuno es corto, la glucemia se mantiene principalmente a partir de la liberación de los depósitos de glucógeno (glucogenolisis). El glucógeno almacenado en hígado y riñón sería capaz, por sí mismo, de mantener la glucemia dentro del rango normal durante, al menos, 4-6 horas en el lactante pequeño, y durante más de 8 horas en niños mayores (Fig. 1).
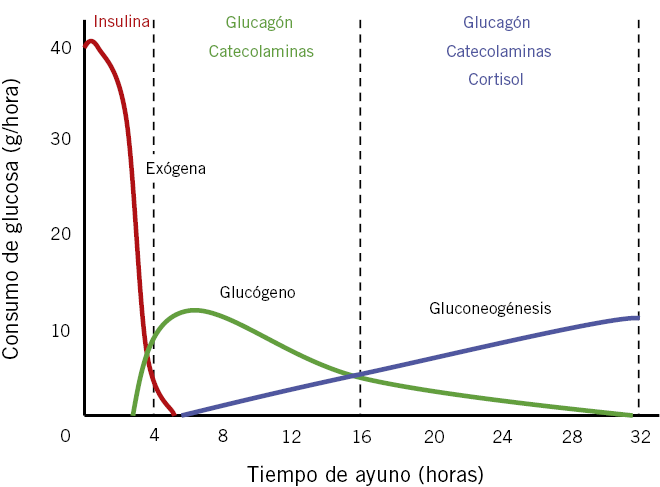
Figura 1. Fases de la homeostasis de la glucosa durante el ayuno (adaptado de Ruderman, et al., 1976). El consumo de glucosa refleja las necesidades medias de un adulto. La duración de la segunda fase es tanto más corta cuanto más pequeño es el niño, debido al menor tamaño de los depósitos hepáticos de glucógeno.
La glucogenolisis es complementada por la gluconeogénesis a partir de los aminoácidos liberados en el recambio proteico (especialmente alanina y glutamina, cuyos niveles séricos se incrementan).
• Si el ayuno se prolonga más de 8-16 horas en un niño normal (24-36 horas en un adulto), los depósitos de glucógeno se van agotando y la gluconeogénesis asume el principal papel en el mantenimiento de la glucemia (fase de ayuno prolongado). Para evitar una excesiva pérdida de proteínas, el tejido adiposo se convierte en la fuente principal de energía para el organismo. Los triglicéridos acumulados en él se descomponen en glicerol (sustrato para la gluconeogénesis) y en ácidos grasos libres (AGL) de cadena larga. Los AGL son oxidados en las mitocondrias hepáticas (β -oxidación) para generar la energía necesaria para la gluconeogénesis, y también en otros tejidos, como el músculo esquelético y cardíaco, contribuyendo al ahorro de glucosa. La β -oxidación de los AGL produce acetil-CoA, que, en ausencia de carbohidratos, no puede ser reciclado a través del ciclo de Krebs y se acumula activando la cetogénesis y generando los cuerpos cetónicos (β -hidroxibutírico y acetoacético), que son utilizados como fuente alternativa de energía por numerosos órganos, contribuyendo también a aumentar la disponibilidad de glucosa para los órganos glucodependientes. A diferencia de los AGL, los cuerpos cetónicos y el lactato atraviesan la barrera hematoencefálica y pueden ser utilizados directamente por las neuronas para obtener energía. De este modo, la producción de cuerpos cetónicos durante el ayuno puede considerarse un mecanismo adaptativo que permite minimizar el consumo de glucosa, evitando así un excesivo catabolismo muscular y lipídico. En los niños pequeños, la presencia de cuerpos cetónicos se observa tras solo 12-18 horas de ayuno; mientras que, en niños mayores, puede no aparecer hasta pasadas 18-24 horas.
Los mecanismos que mantienen la homeostasis de la glucosa en el niño, fuera del período neonatal y una vez que se ha completado la transición entre la vida intra y extrauterina, son esencialmente idénticos a los del adulto. En adultos sanos, la glucemia se mantiene prácticamente en rango normal incluso durante períodos de ayuno de varias semanas; por el contrario, los lactantes y niños pequeños son incapaces de mantener la glucemia tras ayunos relativamente cortos (24-36 horas). La principal diferencia entre lactantes, niños y adultos radica en el ritmo de utilización de la glucosa y en el volumen de sus depósitos neoglucogénicos (Tabla I); de forma que, cuanto más pequeño es el niño, mayor es su consumo proporcional de glucosa, menores sus depósitos de precursores neoglucogénicos y más vulnerable su cerebro a la hipoglucemia. Esta mayor vulnerabilidad del tejido cerebral es especialmente marcada durante los dos primeros años de vida, cuando su crecimiento y desarrollo son mayores y más rápidos.

Homeostasis de la glucosa en el periodo fetal
La glucemia fetal depende fundamentalmente de la GP materna a través de su transferencia placentaria mediante un mecanismo de difusión facilitada a favor de gradiente no regulado por insulina.
Los GLUTs placentarios, incluido el más importante, el GLUT1(2), no son regulados por la insulina y la glucemia fetal depende, fundamentalmente, de la GP materna a través de su transferencia placentaria mediante un mecanismo de difusión facilitada a favor de gradiente de concentración. El resultado es una correlación lineal entre la GP materna y fetal; de forma que, esta última se mantiene alrededor de 60-80 mg/dL, unos 9 mg/dL por debajo de la glucemia materna (70-90 mg/dl)(10). Se cree que la insulina materna, que no atraviesa la barrera placentaria, regularía la GP fetal a través de la GP materna; mientras que, la insulina fetal tendría un papel más relevante en la regulación de los procesos de crecimiento y favorecería el acúmulo de depósitos de energía en el feto, en forma de glucógeno y grasa(10).
En la segunda mitad de la gestación, la secreción de grandes cantidades de lactógeno placentario, progesterona y estrógenos genera en la madre una marcada insulinorresistencia, incrementando la disponibilidad fetal de sustratos maternos (glucosa, aminoácidos y lípidos), lo que favorece el rápido incremento fetal de peso(2). Una de las consecuencias de este aumento del anabolismo fetal es el incremento progresivo de los depósitos de glucógeno hepático, especialmente a partir de la 36 semana de edad gestacional (EG), que pasarían de unos 25 mg/g de hígado, a las 17 semanas de EG, a unos 50 mg/g en el recién nacido (RN) a término. La reducción del flujo placentario puede condicionar un retraso de crecimiento intrauterino (RCIU) y un bajo peso para la EG (BPEG), con disminución de los depósitos de glucógeno, especialmente si el parto se produce prematuramente, lo que favorecería la hipoglucemia perinatal.
Homeostasis de la glucosa en el periodo perinatal(2,10,11)
Los niveles de glucemia, en los RN normales y en las primeras horas de vida, caen a 55-65 mg/dl y en un 30% por debajo de 50 mg/dl, probablemente como resultado de un hiperinsulinismo leve y transitorio, y tienden a normalizarse en 1-3 días (70-100 mg/dl). Es lo que se conoce como “hipoglucemia transicional” y debe diferenciarse de otras causas patológicas de hipoglucemia transitoria o persistente.
Inmediatamente después del nacimiento, la interrupción de la transferencia materna de glucosa obliga al RN a movilizar sus depósitos de glucosa y a ajustar su secreción de insulina a sus niveles de glucemia. Todo ello en un contexto de aportes intermitentes de leche que, además, en los lactados al pecho, contiene en los primeros días mucha grasa y proteínas (calostro), pero escasa cantidad de hidratos de carbono. El resultado es que la glucemia del RN cae en las 2 primeras horas de vida a 55-65 mg/dl (unos 25-30 mg/dL); posteriormente, tiende a aumentar lentamente hasta alcanzar, en 1-3 días, la concentración normal en lactantes, niños y adultos; es decir, alrededor de 70-100 mg/dL. Los niveles más bajos de glucemia se dan en las primeras horas postparto y, como consecuencia, alrededor de un 30% de los RN normales presentarán glucemias < 50 mg/dL en las primeras 24 horas de vida, pero solo un 0,5% después de pasado este periodo(2). Esta caída en los niveles de glucemia en las primeras horas de vida en los RN normales es lo que se conoce como “hipoglucemia transicional” y debe diferenciarse de otras causas patológicas de hipoglucemia transitoria o persistente.
La etiopatogenia de la hipoglucemia transicional no está plenamente aclarada. Los estudios disponibles sugieren que sería una forma leve y transitoria de hiperinsulinismo (hipoglucemia hipocetósica), debido a una disminución en el dintel de GP necesario para suprimir la secreción de insulina en la célula β , 50-65 mg/dL en vez de los 80-85 mg/dl de los niños mayores y adultos(10). Se ha postulado que sería debido a la persistencia transitoria de un mecanismo adaptativo fetal desarrollado para mejorar el crecimiento intraútero. El dintel de GP más bajo en el feto que en la madre permitiría a este mantener su secreción basal de insulina y un crecimiento normal, cuando la glucemia materna, y con ella la fetal, bajan como consecuencia del ayuno o de la disminución de nutrientes. En estas situaciones, el cerebro fetal dispondría, como nutriente alternativo a la glucosa, de abundantes cuerpos cetónicos procedentes de la cetogénesis materna, mucho más acelerada que en mujeres no gestantes.
Situaciones patológicas, como: asfixia perinatal, retraso de crecimiento intrauterino, toxemia materna o eritroblastosis fetal, podrían agravar y prolongar días, semanas e incluso meses, este mecanismo adaptativo, de forma que podría llegar a requerir tratamiento farmacológico.
Concepto de hipoglucemia
Pese a las dificultades en la definición de hipoglucemia(12), en la práctica clínica, existe un cierto consenso en considerar como hipoglucemia un valor de GP ≤ 46 mg/dL(11), a cualquier edad por encima de las 48-72 horas de vida (periodo transicional). En pacientes con DM, la hipoglucemia suele definirse por valores ≤ 70 mg/dL(13).
El primer problema que se plantea a la hora de orientar el diagnóstico de una hipoglucemia es su propia definición que, desde hace años, es objeto de controversia(12). Podríamos definir clínicamente la hipoglucemia, como: una GP lo suficientemente baja como para causar alteración de la función cerebral. Sin embargo, esta definición, como cualquier otra, plantea numerosos problemas:
• Los síntomas de la hipoglucemia son inespecíficos y difíciles de reconocer, y, por otro lado, un valor de GP disminuido puede ser un simple artefacto. Por ello, las guías clínicas de diagnóstico y tratamiento de la hipoglucemia en adultos(14) enfatizan la importancia diagnóstica de documentar la llamada tríada de Whipple: 1) presencia de sintomatología consistente con hipoglucemia; 2) hallazgo de niveles de GP disminuidos; y 3) resolución de los signos y síntomas cuando la GP se normaliza. En los niños, la posibilidad de documentar la tríada de Whipple pocas veces es factible, debido a una sintomatología todavía más inespecífica y a la limitada-ausente capacidad para reconocer y comunicar los síntomas, especialmente en los más pequeños.
• No es posible establecer un punto de corte que delimite lo que es una GP normal-anormal o un nivel de GP por debajo del cual se produzca daño neurológico. Ello se debe a que la respuesta cerebral a la hipoglucemia se produce en un rango relativamente amplio de GP (continuum) y este rango puede verse alterado por la presencia de sustratos alternativos (cuerpos cetónicos, lactato) o por antecedentes recientes de hipoglucemia (hipoglucemias repetidas)(12). Por otro lado, la posibilidad de ocasionar daño cerebral depende no solo del valor de la GP, sino también de otros factores, como: duración, gravedad o, lo que es más importante, coexistencia de otros potenciales agentes lesivos, como la hipoxia o la isquemia.
• La interpretación de una determinación de GP puede estar sujeta a potenciales artefactos o errores en la técnica: consumo de glucosa por tiempo excesivo en procesar la muestra, extracción de vías sin adecuado lavado, etc. Uno de los errores más frecuentes es considerar como fiables los resultados obtenidos mediante glucómetros o reflectómetros portátiles. La utilización de estos aparatos puede conllevar errores de distinto tipo: 1) técnicos, por fallos del aparato, de la metodología empleada o por la utilización de muestras escasas o contaminadas (desinfectantes y agua); 2) de fiabilidad, estos aparatos suelen tener un rango de error en torno al 10-15%, que puede ser mucho mayor cuando la glucemia es baja o alta; y 3) por el tipo de muestra empleado (sangre total y no plasma). La glucemia en sangre total (capilar, arterial o venosa) es un 10-15% más baja que la plasmática, debido al bajo contenido en glucosa de los eritrocitos, por lo que, el rango de error puede aumentar en situaciones de policitemia o anemia. Pese a todas estas posibles fuentes de error, la determinación de la glucemia con estos dispositivos a la cabecera del enfermo posee una incuestionable utilidad derivada de la rapidez de resultados; si bien, cualquiera valor de glucemia por debajo de 60 mg/dL, debería ser confirmado mediante la determinación en laboratorio de la GP y valorar tratamiento si el valor es confirmado. Valores por debajo de 50 mg, especialmente si son sintomáticos, deben recibir tratamiento urgente a la espera de la confirmación por el laboratorio mediante la administración de glucosa oral o intravenosa.
La rapidez para determinar la glucemia en la cabecera del paciente, mediante reflectómetros portátiles de los empleados habitualmente en el control de la diabetes, posee una incuestionable utilidad diagnóstico-terapéutica, pero sus múltiples fuentes de error conllevan que cualquiera valor de glucemia por debajo de 60 mg/dL, debería ser confirmado mediante la determinación en laboratorio de la GP.
Pese a las dificultades en la definición de hipoglucemia, en la práctica clínica, existe un cierto consenso en considerar como hipoglucemia y, por tanto, criterio para iniciar estudios más complejos, un valor de GP ≤ 46 mg/dL (≤ 2,6 mmol/L)(11), a cualquier edad por encima de las 48-72 horas de vida (periodo transicional). Este valor es el que mejor predice la aparición de signos neurofisiológicos de disfunción cerebral, independientemente de que el paciente muestre o no síntomas neuroglucopénicos. En pacientes con diabetes mellitus y en la práctica clínica, suele utilizarse como dintel para iniciar el tratamiento de la hipoglucemia valores ≤ 70 mg/dL (≤ 3,9 mmol/L)(13), dado el mayor riesgo que presentan de hipoglucemia grave y que no es deseable, de cara a una mayor estabilidad glucémica, que se pongan en marcha los mecanismos contrarreguladores.
Concepto de hipoglucemia en el periodo transicional
La definición e interpretación de la hipoglucemia en las primeras 48-72 horas de vida es todavía más compleja y controvertida, especialmente en los prematuros; ya que, el hallazgo de valores de GP por debajo de 30 mg/dl de GP es relativamente frecuente y podría representar un mecanismo de adaptación fisiológica. Más aún, no está claro si el cerebro del RN tiene mayor o menor susceptibilidad a la hipoglucemia que en edades posteriores(7), ni si la hipoglucemia en este periodo es responsable o no de alteraciones posteriores en el neurodesarrollo(4,15).
El hecho estadístico es que, aproximadamente, un 50% de los RN con riesgo de hipoglucemia tendrá una GP< 46 mg/dL y un 19% <36 mg/dL, en la mayoría de los casos en las primeras 24 horas(11), y ello pese a que se promueva una alimentación temprana y frecuente. Por todo ello, no existe un consenso internacional que establezca qué niveles de GP requieren intervención en el RN, ni cuándo ni cómo debe realizarse el cribado de hipoglucemia en los pacientes de riesgo (Tabla II)(7).

Un consenso de expertos ha propuesto utilizar, más que una cifra determinada de GP para definir la hipoglucemia en todos los neonatos (p. ej.: 46 mg/dL), dinteles operacionales que faciliten las decisiones terapéuticas a adoptar ante una determinada situación (hipoglucemia sintomática, asintomática, edad gestacional…)(16). Esta orientación pragmática, a la espera de datos fiables que permitan establecer la relación o no de la hipoglucemia neonatal con la alteración del neurodesarrollo, permitiría evitar investigaciones e intervenciones innecesarias(16-18). A modo de ejemplo, el protocolo de la Sociedad de Pediatría Canadiense, recomienda:
- Realizar el screening de hipoglucemia en pacientes de riesgo a las 2 horas del nacimiento y, posteriormente, cada 3-4 horas hasta que se considere que el riesgo ha disminuido.
- Intervenir cuando la GP sea <32 mg/dL, administrando glucosa i.v., o < 46 mg/dL, administrando suplementos orales(11).
Manifestaciones clínicas
Las manifestaciones clínicas de la hipoglucemia varían, dependiendo, entre otros factores, de la edad del niño, pero son, en general, inespecíficas(1,2,9,19). Esta inespecificidad es tanto mayor cuanto menor es la edad del niño, pudiendo llegar a confundirse en los RN y lactantes pequeños con cuadros clínicos de sepsis o insuficiencia cardiaca.
La semiología de la hipoglucemia (Tabla III) se divide clásicamente en dos categorías: autonómica y neuroglucopénica:
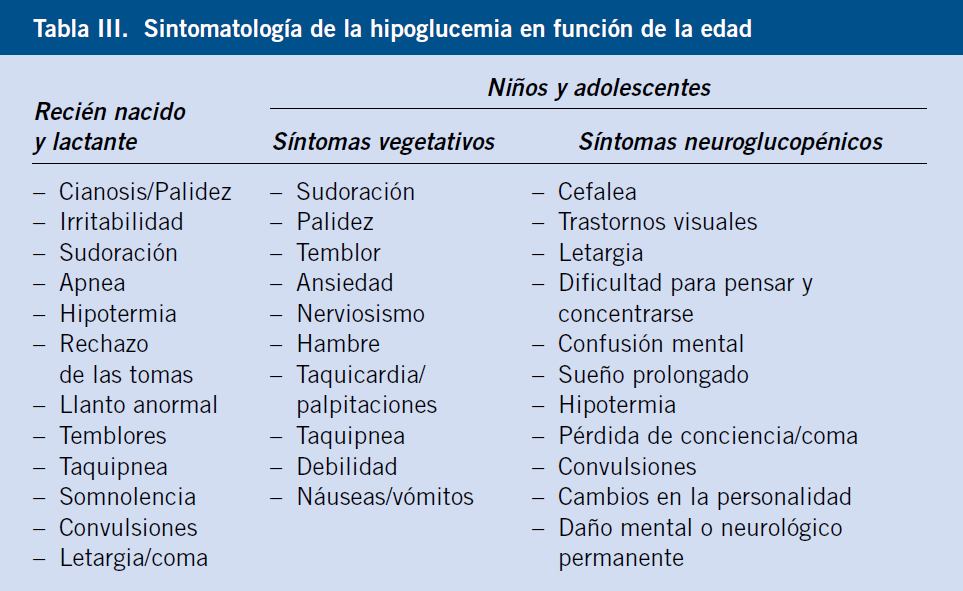
• Las manifestaciones clínicas autonómicas resultarían de la activación, en respuesta a la hipoglucemia, del sistema nervioso simpático y de la médula suprarrenal, que van a liberar: adrenalina, noradrenalina (taquicardia, temblor, ansiedad…) y, en menor medida, acetilcolina (sudoración, hambre…). Suelen preceder a las manifestaciones neuroglucopénicas; por lo que, se consideran una “sintomatología de alarma”. Los síntomas suelen ser más evidentes en hipoglucemias postprandiales y, normalmente, aparecen con glucemias relativamente elevadas (70-40 mg/dL). Si la glucemia sigue descendiendo (50-20 mg/dL) o la hipoglucemia se instaura de manera más lenta y progresiva, aparecen los síntomas neuroglucopénicos.
• Las manifestaciones neuroglucopénicas resultarían de la falta de glucosa en el sistema nervioso central y pueden oscilar, desde síntomas muy sutiles (falta de concentración, irritabilidad…) a convulsiones, coma e incluso la muerte. En los niños mayores, la hipoglucemia grave puede, en ocasiones, inducir déficits neurológicos transitorios de mecanismo etiopatogénico incierto (vasoespasmo cerebral, convulsión focal…), como es el caso de: hemiplejia, hemiparesia (parálisis de Todd), parálisis de pares craneales, etc.
La hipoglucemia puede, también, ser prácticamente asintomática, especialmente cuando la reiteración de episodios de hipoglucemia hace que la percepción por el paciente de los síntomas autonómicos de alarma disminuya (hipoglucemias inadvertidas o hipoglucemias asociadas a fallo autonómico); de forma que, las únicas manifestaciones de la hipoglucemia grave sean las neuroglucopénicas. Este fenómeno es especialmente frecuente, pero no específico, de la reiteración de hipoglucemias en el diabético(5). No obstante, incluso un episodio aislado de hipoglucemia moderada o grave puede bloquear o disminuir el dintel de activación de la respuesta neuroendocrina (activación simpática y hormonas contrarreguladoras) durante 24 horas o más. Por el contrario, en la hiperglucemia crónica, el dintel de glucosa para la respuesta contrarreguladora aumenta(9); de forma que, los síntomas autonómicos pueden aparecer con niveles de glucemia normales.
Etiopatogenia
El mantenimiento de la euglucemia depende de la integridad de un complejo sistema en el que intervienen numerosos elementos(1,11): 1) la ingesta de carbohidratos y su correcta absorción desde el tubo digestivo; 2) la normalidad funcional del hígado, incluyendo la de las enzimas involucradas en la síntesis y la degradación de glucógeno, en la gluconeogénesis, en la β -oxidación de los ácidos grasos y en la cetogénesis; 3) la disponibilidad de sustratos neoglucogénicos endógenos (aminoácidos, glicerol y lactato); y 4) el control óptimo de los procesos anteriores por parte del sistema neuroendocrino. Una alteración en cualquiera de estos elementos puede ser responsable de la aparición de hipoglucemia persistente, por lo que el diagnóstico diferencial de las causas de la misma es extenso y complejo (Tabla IV).

En el periodo neonatal(2,7,10) son frecuentes y características las hipoglucemias transitorias; es decir, aquellas que se resuelven en la primera semana de vida. Son debidas, en muchos casos, a bajos depósitos de glucógeno y de sustratos para la gluconeogénesis (prematuridad, restricción del crecimiento intrauterino) y a la inmadurez de los mecanismos de adaptación al ayuno (pobre desarrollo de la gluconeogénesis y cetogénesis), pero también, como ya ha sido comentado, a hiperinsulinismos transitorios(10), asociados, entre otros factores a: diabetes gestacional o estrés perinatal (Tabla II). No comentaremos más ampliamente estas formas transitorias, porque la gran mayoría de ellas se resuelven espontáneamente en pocos días, necesitando, en ocasiones, de suplementos orales o intravenosos de glucosa; no obstante, en algunos casos, en especial los asociados a hiperinsulinismos, pueden persistir durante semanas o incluso meses(19). Afortunadamente, la mayoría de estos hiperinsulinismos transitorios responden bien a la terapia médica con diazóxido. La utilización de glucocorticoides en RN con hipoglucemia persistente de etiología incierta está desaconsejada.
La etiopatogenia de las hipoglucemias persistentes en la infancia varía con la edad(1,19), de forma que: 1) en los 2 primeros años de vida, los hiperinsulinismos son la primera causa de hipoglucemia, seguidos de los déficits enzimáticos y deficiencias de hormonas contrarreguladoras (hipopituitarismos); 2) entre los 2 y los 8 años, la causa más frecuente es la hipoglucemia cetósica idiopática de la infancia; y 3) en mayores de 8 años, dejando aparte fracasos hepáticos o intoxicaciones, los adenomas/insulinomas pancreáticos, asociados o no a la neoplasia endocrina múltiple tipo 1.
A continuación, intentaremos describir, de forma somera, las causas más importantes de hipoglucemia y sus mecanismos etiopatogénicos.
Hiperinsulinismos congénitos persistentes (HICP)
El HICP es la causa más común y más grave de hipoglucemias reiteradas en el período neonatal y una causa importante de hipoglucemia en edades posteriores.
Su frecuencia varía mucho dependiendo de la población estudiada, oscilando entre 1:20.000 y 1:50.000 RN vivos; si bien, dado el marcado predominio de las formas de herencia autosómico-recesiva (AR), la incidencia puede ser mucho mayor (1:2.500) en comunidades cerradas con alto grado de consanguinidad(1,20).
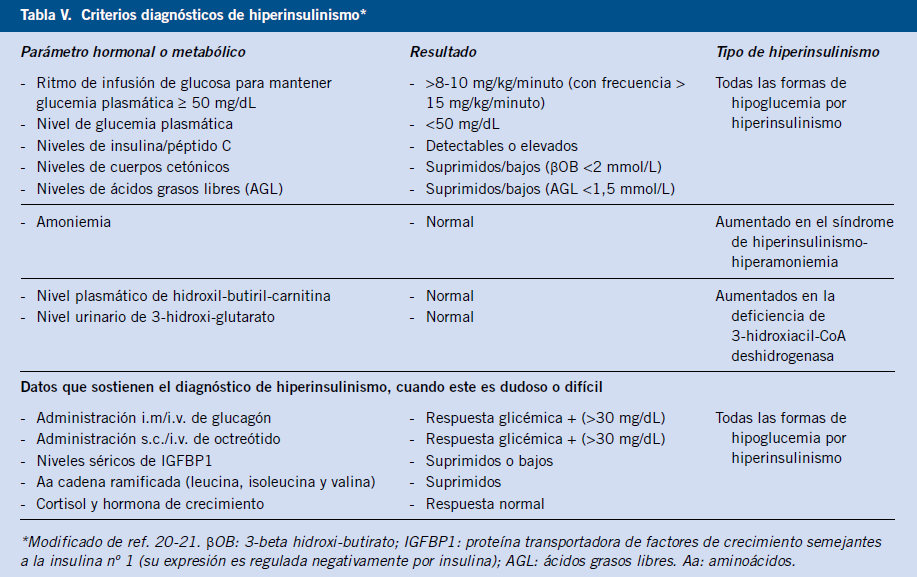
El término de “hiperinsulinismo” indica la liberación inapropiada de insulina para el nivel de glucemia. El diagnóstico se establece ante la presencia de niveles detectables de insulina (>2-3 µUI/ml) o péptido C durante un episodio de hipoglucemia espontáneo o inducido (test de ayuno) (Tabla V).
La gravedad de las hipoglucemias asociadas a HICP reside en que el exceso de insulina limita todas las posibles fuentes energéticas (glucosa, cuerpos cetónicos, ácido láctico y AGL), favoreciendo, más que ninguna otra forma de hipoglucemia, el daño cerebral.
Alrededor del 60% de los HICP se manifiestan en la primera semana de vida, incluyendo las formas más graves que, con frecuencia, asocian también macrosomía fetal, debido al efecto estimulador de la hipersecreción fetal de insulina intraútero. Las formas más leves o moderadas tienden a presentarse con la característica sintomatología inespecífica del niño pequeño (convulsiones, letargia, retraso de desarrollo…) durante el primer año de vida, pero pueden hacerlo más tarde, a veces incluso en la edad adulta.
En los últimos años, se ha producido un considerable avance en el conocimiento de los mecanismos fisiopatológicos que regulan la secreción de insulina y de las bases genéticas de los HICP(21,22); pese a ello, solo en alrededor del 50% de los casos, la base molecular es conocida (Tabla IV).
Desde el punto de vista histológico(20), se distinguen tres formas mayores de HICP: difusas, focales y atípicas. El interés de esta clasificación radica en que tanto las formas focales como las atípicas pueden ser susceptibles de curación mediante pancreatectomía parcial; mientras que, las formas difusas requerirán tratamiento médico prolongado o pancreatectomía total en caso de falta de respuesta al tratamiento médico.
Las anomalías más frecuentes y más graves son las mutaciones inactivantes que afectan a los genes ABCC8 (subunidad SUR1) y KCNJ11 (subunidad Kir6.2), ambos localizados en 11p15.1, que regulan la expresión del canal de potasio dependiente de ATP (KATP) de la célula β (canalopatías). La mayoría y, además, las de mayor severidad y precocidad (inicio en periodo neonatal) son debidas a mutaciones inactivantes, de herencia AR (homocigosis o heterocigosis compuesta), en uno de estos dos genes (Tabla IV), que habitualmente no responden a diazóxido, el fármaco considerado de primera elección en HICP. Las mutaciones inactivantes en ABCC8 o KCNJ11 de herencia autosómica dominante (AD) suelen causar formas de HICP menos graves y de inicio más tardío y suelen, aunque no siempre, responder a diazóxido.
Las mutaciones germinales AR o AD en los genes ABCC8 y KCNJ11 dan lugar a formas histológicas difusas. Las formas focales afectan a áreas pequeñas y pobremente delimitada del páncreas (≈ 2-10 mm de diámetro) que pueden presentar tentáculos o áreas satélites próximas, lo que exige, en caso de tratamiento quirúrgico, unos bordes relativamente amplios de extirpación para evitar recurrencias.
La herencia de las formas focales suele ser esporádica y el mecanismo genético conlleva dos eventos independientes: 1º) una mutación germinal en el alelo paterno de ABCC8 o KCNJ11; 2º) una pérdida somática del alelo materno 11 p (11p15.1-11p15.5) en la región de la lesión focal, lo que condiciona una disomía uniparental paterna para una serie de genes improntados y localizados en esa área cromosómica (genes tumorales supresores maternos H19 y CDKN1C, y del gen paterno IGF2), responsable, probablemente, de la hiperplasia focal adenomatosa del clon de células β afectadas. Más recientemente, han sido descritas las formas focales atípicas, caracterizadas por la presencia en el páncreas de dos tipos de células que coexisten (mosaicismo histológico), unas grandes con citoplasma y núcleo grandes y otras pequeñas con citoplasma y núcleo pequeño. Las células grandes son hiperactivas y tienden a confinarse en uno o pocos lóbulos, lo que las hace susceptibles de extirpación (pancreatectomía parcial) y curación del HICP. Esas células grandes presentan una hipersecreción de insulina con dintel bajo de glucosa, con sobrexpresión en la mayoría de los casos de hexoquinasa 1.
La segunda causa más común de HICP son las mutaciones con ganancia de función en GLUD1, el gen que codifica para la glutamato-dehidrogenasa (GDH). Esta enzima modula la secreción de insulina inducida por aminoácidos. Se heredan de forma AD, aunque el 80% son mutaciones de novo. El cuadro clínico se caracteriza por episodios repetidos de hipoglucemia moderada-grave que, a veces, por su inespecificidad clínica, no se reconocen hasta los 1-2 años, y que se producen, con frecuencia, 30-90 minutos después de la ingestión de proteínas (hipoglucemia sensible a proteínas) y acompañadas de ligera hiperamoniemia (60-150 µmol/l)(23). Las HICP por mutaciones activadoras en GLUD1 suelen responder positivamente a diazóxido. La hiperamoniemia no requiere tratamiento, pero da nombre a la enfermedad: “síndrome de hiperinsulinismo-hiperamoniemia”.
Hiperinsulinismos adquiridos
Las hipoglucemias, debidas a un exceso relativo de insulina, son la complicación aguda más frecuente de la diabetes mellitus(DM)(13). La mayoría de los episodios de hipoglucemia suelen ser aislados y leves, sin afectación de la conciencia. En estos casos, el tratamiento consistirá en reposo y administración de suplementos de hidratos de carbono, 10-20 gramos, de absorción rápida, en forma de: glucosa, azúcar, zumos, etc. Se deben esperar, al menos, 15 minutos antes de volver a medir la glucemia para evitar la sobrecorrección y la hiperglucemia posterior. Si va a transcurrir más de una hora antes de la siguiente comida planificada, el paciente debe ingerir otros 10-20 g de hidratos de carbono de absorción más lenta (pan, leche, fruta). Si la hipoglucemia es grave, con afectación del nivel de conciencia (coma, incoherencia, desorientación, convulsiones, etc.), el aporte oral de glucosa está contraindicado por el riesgo de aspiración, pero puede inducirse la liberación endógena de glucosa a partir de los depósitos de glucógeno hepático mediante la administración de glucagón parenteral (0,1-0,3 mg/kg por vía s.c., i.m. o i.v.; dosis máxima de 1 mg). En medio hospitalario y ante una hipoglucemia grave lo ideal es administrar glucosa i.v al 10% (2 ml/kg) y, posteriormente, suero glucosado al 10%. La administración de glucagón i.m. puede también ser de utilidad en estos casos si se presentan dificultades en la obtención de una vía. Si las hipoglucemias son graves o reiteradas, será necesario modificar la pauta habitual de tratamiento, al objeto de prevenir nuevos episodios.
Dejando aparte la DM, los insulinomas son la causa más frecuente de hipoglucemia por hiperinsulinismo persistente en el niño mayor y en el adulto, aunque pueden presentarse a edades muy precoces(24). En el niño, suelen ser de pequeño tamaño y de difícil localización, generalmente esporádicos, pero ocasionalmente asociados a neoplasia endocrina múltiple de tipo 1 (MEN1). Los insulinomas pueden, en ocasiones, responder a diazóxido, pero el tratamiento definitivo consistirá en la extirpación del adenoma y si este no es localizable con las diferentes técnicas de imagen, pancreatectomía distal subtotal.
La hipoglucemia puede ser también el resultado de la administración errónea o en el contexto de un síndrome de Munchausen por poderes de insulina o hipoglucemiantes orales (sulfonil-ureas). El diagnóstico de un síndrome de Munchausen por poderes requiere de un alto índice de sospecha; ya que, las sulfonil-ureas remedan perfectamente un hiperinsulinismo endógeno (aumento de insulina y péptido C) y los nuevos análogos de insulina no son detectados en los inmunoensayos convencionales de insulina (patrón clínico-metabólico sugerente de hiperinsulinismo, pero con insulina y péptido C indetectables).
La hipoglucemia autoinmune (síndrome de Hirata)(25) es una causa excepcional de hipoglucemia, salvo en Japón y Corea. Es una hipoglucemia, habitualmente de aparición postprandial, que puede ocurrir a cualquier edad, aunque es excepcional en la edad pediátrica, y resultaría de la presencia de anticuerpos dirigidos contra la insulina (insulina muy elevada y péptido C elevado) o el receptor de insulina (insulina y péptido C bajos). Se asocia a determinados fenotipos HLA y a enfermedades autoinmunes, y parece ser desencadenado, en la mayoría de los casos por fármacos, sobre todo, portadores del grupo sulfidrilo (metimazol, carbimazol, hidralacina…). Suele ser transitoria (meses) y normalizarse tras la retirada del fármaco, y suele responder a tratamiento dietético (comidas frecuentes), aunque en ocasiones, la plasmaféresis o el tratamiento esteroideo pueden ser necesarios.
La hipoglucemia hiperinsulinémica postprandial se produce 1-2 horas tras la ingesta, como consecuencia de una secreción inapropiada de insulina en respuesta a la comida. La causa más frecuente es un síndrome de dumping tras cirugía (funduplicatura de Nissen)(26). En estos casos, se recomiendan los carbohidratos complejos, evitando los de absorción rápida, y suplementos de acarbosa en las comidas (12-75 mg/dosis). También, se han descrito situaciones de hipoglucemias, denominadas reactivas, con sintomatología sugerente de hipoglucemia, 2-4 horas tras la comida, de etiología desconocida. Son frecuentes en niñas adolescentes con historia familiar positiva de síntomas similares en la madre. El diagnóstico conlleva la confirmación de la hipoglucemia y descartar otras posibles causas, como el hiperinsulinismo.
Deficiencia de hormonas contrainsulares
Entre un 10 y un 25% de los hipopituitarismos, bien por deficiencia aislada de hormona de crecimiento o de hormona adrenocorticotropa, o bien por deficiencia hipofisaria múltiple, se asocian con hipoglucemia por disminución de la gluconeogénesis. La presencia de defectos morfológicos de línea media (labio leporino, paladar hendido, displasia septo-óptica…) e hipoglucemia, debe hacer sospechar hipopituitarismo. La supresión iatrogénica de la secreción de cortisol puede, en situaciones de estrés intercurrente, provocar vómitos, hipotensión e hipoglucemia (insuficiencia suprarrenal aguda). La asociación de hipoglucemia e hiperpigmentación cutánea debe orientar hacia situaciones de insuficiencia suprarrenal primaria (enfermedad de Addison). El tratamiento en todas estas situaciones consistiría en la sustitución de la hormona u hormonas deficitarias.
Errores innatos del metabolismo (EIM)(2,7,11,19,27,28)
La mayoría de las enfermedades metabólicas son de herencia AR; por lo que, son extremadamente infrecuentes excepto en situaciones de consanguinidad. Pueden producir hipoglucemia por mecanismos muy variados y algunas de ellas están incluidas en los programas de detección de enfermedades metabólicas del cribado neonatal.
EIM que afectan al depósito o liberación de glucógeno (glucogenosis)
Las glucogenosis (GSD: Glycogen Storage Disease), junto con el hiperinsulinismo y el hipopituitarismo, son las causas principales de hipoglucemia en el período neonatal, aunque la mayoría de ellas se presentan más tardíamente. Como la mayoría de los EIM, son enfermedades raras (incidencia combinada de ≈ 1:20.000 RN vivos) y la mayoría de herencia AR. Los tipos de GSD que producen hipoglucemia son aquellos que tienen una afectación fundamentalmente hepática, y lo hacen habitualmente tras 3-12 horas de ayuno. Además del déficit de glucógeno-sintetasa hepático (GSD 0), se incluyen: GSD I (déficit de glucosa 6 fosfatasa), GSD III (déficit de la enzima desrramificante, amilo-1-6-glucosidasa), GSD VI (déficit de fosforilasa hepática) y GSD IX (deficiencia de fosforilasa quinasa). Salvo la GSD 0, que además es extremadamente rara (solo se han descrito algunos casos aislados), el resto de GSD hepáticas se acompañan de marcada hepatomegalia.
El tipo I de GSD (GSD1) es la forma más frecuente y grave de GSD, y se debe a la deficiencia de la enzima que hidroliza la glucosa 6 fosfato a glucosa libre (GSD1a) o de su transportador microsomal (GSD1b), último paso de la glucogenolisis, pero también es una enzima de la gluconeogénesis. La afectación de ambas vías metabólicas favorece el desarrollo de hipoglucemia persistente y recurrente. En su forma clásica (GSD1a, frecuencia 1:100.000), los pacientes presentan hipoglucemia desde la infancia temprana junto con: hepatomegalia severa, obesidad troncular, xantomas eruptivos (hipertrigliceridemia e hipercolesterolemia), acidosis metabólica (marcada acidosis láctica y moderada cetosis), hiperuricemia, hipofosfatemia, alteraciones de la adhesividad plaquetaria y severo fallo de crecimiento. La hipertransaminasemia generalmente es más moderada que en el resto de los tipos. La sintomatología de la hipoglucemia suele ser escasa y los pacientes exhiben una alta tolerancia a la hipoglucemia crónica, reflejando la adaptación crónica cerebral a fuentes alternativas (láctico). En el tipo GSD1b se asocia, además, neutropenia y anomalías funcionales del neutrófilo, que condicionan infecciones bacterianas graves de repetición y úlceras en la mucosa oral e intestinal.
En las GSD tipo III y tipos VI y IX, la hipoglucemia es menos frecuente y la hepatomegalia menos severa. La menor elevación de hormonas contrarreguladoras en respuesta a la hipoglucemia limita la acidosis láctica, la hiperlipemia, la cetonemia y la hiperuricemia. La elevación de las transaminasas suele ser mayor, particularmente en la GSD tipo III. Además, en la GSD tipo IIIa, debido a la afectación muscular, los niveles séricos de creatín-fosfoquinasa (CPK) están elevados.
El diagnóstico definitivo de las GSD debe establecerse mediante estudios moleculares, ante una sospecha clínica y bioquímica compatible, puesto que la mayoría de las mutaciones para cada una de las GSD han sido establecidas. La biopsia hepática con la medición de la actividad enzimática está en desuso y debe reservarse para los casos de diagnóstico incierto(28).
El tratamiento de las GSD consiste básicamente en evitar las hipoglucemias mediante la administración frecuente durante el día de hidratos de carbono (c/2-3 horas), preferentemente en forma de hidratos de carbono complejos, junto con un suplemento continuo de glucosa durante la noche mediante sonda nasogástrica (6-8 mg/kg/min).
EIM que afectan a la gluconeogénesis(27)
Estos EIM se caracterizan por hipoglucemias recurrentes con acidosis láctica y un mayor o menor grado de cetosis. En este grupo, se incluyen los déficits de: piruvato-carboxilasa, fosfoenol-piruvato-carboxiquinasa, fructosa 1-6-bifosfatasa y el tipo 1 de GSD (déficit de glucosa 6 fosfatasa). La hipoglucemia en ayunas es frecuente, debido a la imposibilidad de sintetizar glucosa a partir de precursores carbonados (alanina, piruvato, lactato y glicerol) una vez deplecionados los depósitos de glucógeno.
La deficiencia de piruvato-carboxilasa suele presentarse en los primeros meses de vida con retraso psicomotor y episodios intermitentes de acidosis metabólica. Se caracteriza por un cuadro de acidosis láctica con afectación severa del sistema nervioso central (encefalopatía, convulsiones, hipotonía y coma), que puede acompañarse de hipoglucemia y afectación tubular renal (acidosis tubular). Esta enzima mitocondrial cataliza la formación de oxalacetato a partir de piruvato en presencia de ATP, primer paso en la gluconeogénesis. La disminución/ausencia de oxalacetato bloquea el primer paso de la gluconeogénesis (hipoglucemias de ayuno), interfiere en el correcto funcionamiento del ciclo de Krebs (aumento de lactato, piruvato y otros metabolitos intermedios) y, en las formas más graves, puede provocar, por reducción de los niveles de aspartato, intermediario del ciclo de la urea, acúmulo de citrulina y amonio. La acidosis láctica se incrementa como consecuencia de la interferencia con el ciclo de Cori (intercambio cíclico de glucosa y láctico entre el hígado y el músculo). Bioquímicamente, las formas más graves de presentación neonatal (tipo B) se caracterizan por: acidosis láctica, ratio lactato/piruvato muy elevado (>30; VN: <10), hipercetonemia postprandial, ratio beta-OH-butírico/acetoacético normal o bajo (<1,5), hiperamoniemia, citrulinemia e hiperlisinemia. No existe tratamiento satisfactorio y se recomienda una dieta rica en hidratos de carbono y bicarbonato para compensar la acidosis láctica.
La deficiencia de fosfoenol-piruvato-carboxiquinasa es extremadamente rara y la hipoglucemia aparece durante la infancia asociada a infiltración grasa del hígado (hepatomegalia), riñón y otros órganos debido al incremento en la formación de acetil Co A. Hipotonía, fallo de medro y acidosis láctica son otros hallazgos frecuentes.
La deficiencia de fructosa 1-6 bifosfatasa es una causa rara de hipoglucemia que suele manifestarse en el período neonatal, aunque el 50% lo hace más tardíamente. Transforma la fructosa 1-6 difosfato en fructosa 6 fosfato y es la enzima clave en el proceso de gluconeogénesis, desde lactato, piruvato, alanina y glicerol. Las hipoglucemias suelen ser graves (convulsiones y coma) y presentarse en relación con el ayuno o la presencia de enfermedades intercurrentes, acompañadas de: marcada acidosis láctica, cetosis, hepatomegalia (sin hipertransaminasemia) e hiperuricemia. El diagnóstico se realiza por estudios moleculares o enzimáticos, en biopsia hepática, y el tratamiento consiste en evitar el ayuno prolongado y reducir el aporte de fructosa de la dieta.
EIM que afectan al metabolismo de los ácidos grasos(27-29)
Desde su descripción inicial en 1973, se han identificado más de 20 EIM diferentes que afectan a la β -oxidación de los AGL, todos ellos heredados de manera AR. Los AGL representan en el niño la principal fuente energética (80%) en situaciones de ayuno prolongado (12-24 horas de ayuno); de ahí que, sus defectos suelan hacerse aparentes clínicamente solo con el ayuno o la inanición asociada a enfermedades catabólicas y vómitos. Los estudios metabólicos realizados fuera de estas situaciones pueden ser normales; por lo que, es necesario un alto índice de sospecha para su diagnóstico.
Algunos tejidos, como el hígado y el tejido muscular esquelético, son capaces de catabolizar los AGL de cadena larga (C14 a C20), mediante β -oxidación, hasta CO2 y H2O. En el citoplasma celular, los AGL se acilan con el CoA citoplasmático por acción de la acil CoA sintetasa, formando AGL-acil CoA, que son transportados a la mitocondria, precisando en el caso de los de cadena larga (> 12 carbonos) un sistema de transporte unido a carnitina (ciclo de la carnitina). La β -oxidación mitocondrial tiene como función acortar en dos átomos de carbono los AGL-acil-CoA, generando acetil-CoA, que puede entrar en el ciclo de Krebs para generar la energía necesaria para la gluconeogénesis y, en el hígado, además, en situación de ayuno, puede ser empleado para sintetizar cuerpos cetónicos (cetogénesis: β -hidroxibutírico y acetoacético), que son utilizados, a su vez, como fuente alternativa de energía por numerosos órganos (cetolisis), incluido el cerebro. La falta de síntesis de Acetil-CoA en las alteraciones de la β -oxidación no activa la gluconeogénesis (hipoglucemia) y no se sintetizan cuerpos cetónicos (hipocetosis), fracasando la síntesis de sustratos energéticos e inactivándose el ciclo de la urea (hiperamoniemia) con toxicidad neuronal grave. Un hallazgo diagnóstico característico de estos EIM es un cociente AGL (mMol)/3-OH-butirato (mMol) mayor de 2.
Varias vías metabólicas alternativas adquieren importancia cuando la β -oxidación mitocondrial está alterada. La β -oxidación peroxisomal permite continuar el metabolismo de los AGL de cadena larga (>20 átomos de carbono); mientras que, la ω -oxidación en el citosol conduce a la producción de los característicos ácidos grasos dicarboxílicos, a menudo presentes en estos trastornos. Tanto los AGL-acil CoA como los dicarboxílicos se esterifican con carnitina (acilcarnitinas) y glicina (acilglicinas). El exceso de formación de acilcarnitinas, además de ser tóxico, produce una deficiencia de carnitina libre y un exceso de carnitina esterificada, lo que inhibe el sistema transportador de carnitina y genera una deficiencia de carnitina total. Cada una de las diferentes alteraciones de la β-oxidación genera un perfil de carnitinas y acil carnitinas que puede ser de utilidad en el diagnóstico de estos EIM, orientando los estudios moleculares, que son la base actual del diagnóstico de estas enfermedades.
Las manifestaciones clínicas de los defectos de la β-oxidación pueden ser muy variables, pero obedecen a dos mecanismos básicos: déficit energético y/o intoxicación por la acumulación de determinados metabolitos. El corazón, el músculo esquelético y el hígado son, desde el punto de vista energético, particularmente dependientes de esta vía y la acumulación de determinados metabolitos tóxicos parece ser la responsable de las arritmias, de la afectación neurológica, junto con la hipoglucemia, y, posiblemente también, de los episodios de descompensación fulminante y muerte súbita. Las manifestaciones clínicas afectan especialmente: hígado (hipoglucemia hipocetósica con hiperlactacidemia, hepatomegalia, hiperamoniemia, esteatosis, elevación de transaminasas), miocardio (muerte súbita, arritmias y cardiomiopatía dilatada o hipertrófica), músculo esquelético (hipotonía, miopatía lipídica, rabdomiolisis o elevación de CPK), retina (retinopatía pigmentosa) y cerebro (síndrome de Reye-like, hipsarritmias, leucodistrófia, edema cerebral…).
Aunque los trastornos de la β-oxidación son enfermedades raras, son más frecuentes de lo que se cree, especialmente algunos de ellos, como es el caso de la deficiencia de acil-CoA deshidrogenasa de cadena media (MCAD; metaboliza los acil-CoA de 6-12 átomos de carbono), que afecta aproximadamente a 1:10.000 RN. También, relativamente frecuentes y responsables de hipoglucemia serían: la deficiencia de 3-hidroxiacil-CoA deshidrogenasa de cadena larga (LCAD; acil-CoA de 10-18 átomos de carbono) y la deficiencia de acil-CoA deshidrogenasa de cadena muy larga (VLCAD: acil-CoA de 14-24 átomos de carbono).
Algunas drogas o substancias químicas, como el ácido valproico (VPA), de uso frecuente en Pediatría, pueden interferir con la β-oxidación de los AGL y simular o empeorar estos trastornos congénitos. EL VPA es un ácido graso ramificado, que se metaboliza principalmente por ?-oxidación hepática, siendo la carnitina la responsable de su entrada en la mitocondria. Su uso, por tanto, puede ocasionar una depleción de carnitina. Además, el VPA puede generar una inhibición de la N-acetil-glutamato sintetasa (NAGS), bloqueando o dificultando el flujo normal del ciclo de la urea y empeorando esta situación. Así pues, el VPA puede producir hipoglucemia hipocetósica con síndrome Reye-like, hiperamoniemia y deficiencia secundaria de carnitina. De ahí, la conveniencia, en pacientes en tratamiento con VPA, de vigilar (vómitos, decaimiento o encefalopatía) y tratar una posible deficiencia de carnitina e, incluso, de suplementar sistemáticamente con cartinina a pacientes con dicho tratamiento.
El tratamiento urgente de los errores de la β-oxidación de los AGL (debut y/o descompensaciones) incluye: 1) el aporte de glucosa intravenosa para mantener glucemias entre 100-120 mg/dL; 2) el aporte de bicarbonato para la corrección de la acidosis; 3) la utilización de N-carbamil-glutamato (Carbaglu®), a una dosis inicial de 100 mg/kg, seguida de 100-250 mg/kg/día repartido en 4 dosis, para mejorar la hiperamoniemia; 4) la hemodiafiltración o hemodialisis para limpiar de metabolitos tóxicos; y 5) el aporte de cofactores, carnitina y riboflavina, dependiendo del tipo de defecto de la β-oxidación.
El tratamiento de mantenimiento o de prevención de las descompensaciones incluiría medidas: dietéticas, farmacológicas y sintomáticas. Desde el punto de vista dietético, se deben prevenir los períodos de ayuno (no más de 8 horas), asegurar un aporte calórico suficiente durante los períodos de estrés catabólico y restringir el aporte graso, incrementando el aporte de carbohidratos. En lo que al tratamiento farmacológico se refiere, dependería de la alteración, pero se basa en la utilización de detoxificadores, como la carnitina o la glicina, y de coenzimas, como la riboflavina. Es importante, también, evitar fármacos que consuman carnitina, entre otros: ácido piválico, valproico, salicilatos y paracetamol.
EIM que afectan al metabolismo intermediario y a la homeostasis de la glucosa(27-29)
Varios trastornos del metabolismo de los aminoácidos (acidemias orgánicas) pueden acompañarse de hipoglucemia, como es el caso, entre otros, de: la acidemia metilmalónica (primaria por EIM o secundaria a déficit de vitamina B12), la aciduria 3-hidroxi-3-metil glutárica (un trastorno que también afecta a la β-oxidación de los AGL), la enfermedad de la orina con olor a jarabe de arce o la tirosinemia hereditaria. Todos estos EIM son extremadamente raros y, aunque la hipoglucemia (cetósica o no, dependiendo del trastorno) puede estar presente y mediada por mecanismos fisiopatológicos diferentes, no suele ser la manifestación más llamativa. En general, estos pacientes suelen presentarse con retraso del crecimiento, afectación neurológica grave y vómitos recurrentes, asociados a hiperamoniemia y acidosis metabólica. El diagnóstico suele orientarse a partir de la determinación de ácidos orgánicos en orina y del perfil de aminoácidos en plasma. Posteriormente, la sospecha diagnóstica se confirma, en la mayoría de los casos, mediante estudios moleculares.
La intolerancia a la fructosa es el resultado del déficit de fructosa 1-fosfato aldolasa y las manifestaciones clínicas se inician con la introducción de la fructosa en la dieta, habitualmente por encima de los 4 meses de vida. La ingestión de fructosa provoca: vómitos, dolor abdominal, diarrea crónica, hipoglucemia por inhibición de la gluconeogénesis y de la glucogenolisis (acúmulo de fructosa 1-P), con hiperlactacidemia e hiperuricemia, que puede evolucionar a shock y fallo hepático. La ingestión crónica de pequeñas cantidades de fructosa ocasiona fracaso de crecimiento con fallo hepático, tubulopatía Fanconi-like presencia de cuerpos reductores en orina. Los pacientes tienden a mostrar aversión por los alimentos que contienen fructosa, sacarosa o sorbitol.
En la galactosemia clásica, por deficiencia de galactosa 1-fosfato uridil-transferasa (metaboliza el monosacarido galactosa a glucosa 1-fosfato) la hipoglucemia es infrecuente y las manifestaciones clínicas predominantes son otras: vómitos, diarrea, ictericia, sepsis por E. Colli, fallo de medro, fracaso hepático (hipertransaminasemia, alteración de la coagulación) y renal (tubulopatía proximal) con galactosuria (cuerpos reductores en orina positivos: Clinitest®). Las manifestaciones clínicas se producen en las primeras semanas de vida tras la ingestión de galactosa (lactancia) en el período neonatal y solo aparece hipoglucemia cuando el fracaso hepático es prácticamente terminal. El tratamiento consiste en la supresión de por vida de todas las fuentes de galactosa de la dieta, incluida la lactosa presente en medicamentos y múltiples productos comerciales.
Hipoglucemia cetósica idiopática
Es la causa más común de hipoglucemia en la infancia y se caracteriza por episodios recurrentes de hipoglucemia cetósica en niños pequeños, coincidiendo con las infecciones habituales y la reducción de la ingesta.
En estos niños, un ayuno de 12-24 horas determina la hipoglucemia, que se acompaña de cetonemia y cetonuria intensas. El cuadro clínico se presenta habitualmente entre los 18 meses y los 5 años, remitiendo espontáneamente a los 8-9 años de edad. Es más frecuente en niños con retraso póndero-estatural (masa muscular escasa) y con antecedentes de hipoglucemia transitoria en el período neonatal. Los mecanismos fisiopatológicos subyacentes no están plenamente aclarados y la respuesta hormonal y metabólica a la hipoglucemia es prácticamente normal. El único hallazgo patológico es la marcada disminución, en situación de hipoglucemia, de los niveles séricos de alanina, el principal aminoácido precursor de la gluconeogénesis, cuya formación y liberación desde el músculo, en situaciones de restricción calórica, está aumentada por la presencia del ciclo alanina-glucosa (ciclo de Cahill) y por su formación de novo en el músculo a partir del catabolismo de aminoácidos de cadena ramificada. Por ello, se ha sugerido que el defecto en la hipoglucemia cetósica idiopática radicaría, probablemente, en algunos de los complejos pasos metabólicos implicados en el catabolismo proteico o en la síntesis y liberación de alanina por el músculo. También, se ha sugerido que podría resultar del efecto sinérgico o acumulativo de deficiencias parciales de enzimas implicadas en la homeostasis de la glucosa.
El tratamiento es, principalmente, preventivo, a la espera de la resolución espontánea del cuadro clínico con la edad. Deben evitarse los períodos de ayuno prolongados, sobre todo durante la noche, aportando a última hora hidratos de carbono de absorción lenta. En los períodos de enfermedad intercurrente, deben ofrecerse frecuentemente alimentos líquidos con alto contenido en hidratos de carbono y vigilar en orina la presencia de cuerpos cetónicos, cuya aparición precede en unas horas al desarrollo de la hipoglucemia. Si los aportes orales de hidratos de carbono no son tolerados por el niño, este debe ser ingresado para administración intravenosa de glucosa. Si las hipoglucemias son reiteradas o graves, deben descartarse otras causas de hipoglucemia cetósica, siendo en estos casos, la hipoglucemia cetósica idiopática un diagnóstico de exclusión.
Hipoglucemia asociada a enfermedades graves
La hipoglucemia es frecuente en pacientes con fallo multiorgánico o enfermedad grave, tales como: sepsis, traumatismo craneal severo, fallo cardíaco o renal, necrosis hepática aguda, pancreatitis… La etiopatogenia en estos casos suele ser multifactorial: falta de sustratos, malnutrición, aumento del consumo de glucosa, alteración de la gluconeogénesis, drogas administradas, citoquinas… La malaria grave se asocia específicamente a hipoglucemia como resultado del aumento de consumo de glucosa por el parásito, pero también por el efecto de la medicación que, como la quinina puede estimular la secreción de insulina.
Orientación diagnóstica(1,2,5-7,9,11,19,20,22,30)
Al objeto de evitar estudios innecesarios, solo deben estudiarse las hipoglucemias: en niños mayores con la tríada de Whipple documentada, en niños pequeños con glucemia plasmática (no capilar) < 60 mg/dL y en RN solo a partir de las 48-72 horas de vida (pasada la hipoglucemia transicional)(7).
La sintomatología de la hipoglucemia puede ser muy inespecífica en niños y existe el riesgo de realizar estudios innecesarios en pacientes con falsa sospecha de hipoglucemia. Por ello, la guía de la Sociedad Americana de Endocrinología Pediátrica para el manejo de la hipoglucemia en niños(7) recomienda estudiar solamente:
• A los niños mayores (capaces de explicar sus síntomas) solo si la tríada de Whipple está documentada.
• A los niños más pequeños, con dificultades para expresar y comunicar sus síntomas, solo cuando se haya documentado en laboratorio una GP inferior al dintel normal de respuesta neurogénica (< 60 mg/dL).
• En el caso de RN con alta sospecha de hipoglucemia persistente, realizar evaluaciones diagnósticas solo a partir de las 48-72 horas de vida, cuando el periodo de regulación transicional de la glucosa haya pasado, y solo en aquellas situaciones de riesgo (Tabla II), antes del alta, mediante estudios diagnósticos específicos o asegurando que el niño es capaz de tolerar un ayuno de 6-8 horas (durante el test de ayuno, glucemia ≥ 70 mg/dL).
Averiguar la causa de la hipoglucemia es esencial para planear el tratamiento más adecuado, pero no siempre es sencillo. Se recomienda llevar a cabo una aproximación diagnóstica sistemática y gradual, antes de someter al paciente a las numerosas pruebas diagnósticas disponibles, muchas de las cuales son potencialmente peligrosas y difíciles de interpretar. La anamnesis y la exploración física detalladas permiten sospechar la causa de la hipoglucemia en más del 90% de los pacientes.
Historia clínica
Se deben buscar y recoger antecedentes familiares: de consanguinidad, dada la frecuencia de enfermedades de herencia AR responsables de hipoglucemia; de fallecimientos en la infancia de causa no aclarada (muerte súbita del lactante) o de síndrome de Reye, sugerentes de enfermedad metabólica (EIM no diagnosticados), especialmente de alteraciones de la ?-oxidación de ácidos grasos; de hiperinsulinismos o deficiencias hormonales; y de enfermedades autoinmunes (hiperinsulinismo autoinmune).
La edad de presentación y la relación de los episodios con la ingesta (tiempo de ayuno previo a la aparición de la hipoglucemia y coincidencia temporal con la introducción de ciertos alimentos) son dos datos esenciales para orientar el diagnóstico etiológico:
• El inicio de los episodios de hipoglucemia en el primer año de vida sugerirá, en primer lugar, hiperinsulinismo, pero también EIM o deficiencias hormonales hipofisarias. Pueden presentarse a cualquier edad, pero de no hacerlo en el periodo neonatal, es más frecuente que lo hagan a partir de los 6-7 meses de vida, cuando el lactante realiza una pausa alimentaria nocturna relativamente prolongada y padece numerosas infecciones que dificultan la alimentación. Si los síntomas se inician a partir de los 12-18 meses, el diagnóstico más probable será el de hipoglucemia cetósica idiopática y, con menor frecuencia, hiperinsulinismos tardíos o alteraciones metabólicas leves. Los hiperinsulinismos que aparecen en niños mayores deben hacer sospechar la existencia de un adenoma pancreático.
• Los mecanismos contrarreguladores de la hipoglucemia se van poniendo en marcha de forma secuencial: disminución de la secreción de insulina, glucogenolisis, gluconeogénesis a expensas de aminoácidos y, finalmente, lipólisis (?-oxidación de los ácidos grasos y cetogénesis) con gluconeogénesis a partir de glicerol. De ahí que, la relación de los episodios de hipoglucemia con la ingesta de nutrientes y el tiempo transcurrido desde la última ingesta sean un excelente orientador diagnóstico del defecto subyacente:
- Si la hipoglucemia se desencadena de forma inconsistente con el tiempo de ayuno o en las 2-3 horas siguientes a la ingesta, la causa más probable es el hiperinsulinismo.
- Los episodios de hipoglucemia que aparecen tras la ingesta de alimentos proteicos (hipoglucemia leucín-sensible) sugieren hiperinsulinismo por mutaciones en GLUD1 (síndrome hiperinsulinismo-hiperamoniemia) o en HADH (deficiencias de 3-hidroxiacil-CoA-deshidrogenasa, AR), pero también pueden aparecer en algunas enfermedades metabólicas. Los pacientes con galactosemia pueden presentar hipoglucemia postprandial en los primeros días o semanas de vida (la leche materna y las fórmulas habituales contienen galactosa); mientras que, la intolerancia hereditaria a la fructosa se manifiesta 2-3 horas después de la ingesta de frutas o miel, habitualmente a partir de los 5-6 meses de vida y, a veces, con determinados jarabes que llevan fructosa como excipiente.
- La mayoría de las hipoglucemias en la infancia aparecen tras un cierto tiempo de ayuno. El tiempo de tolerancia al ayuno es corto en la glucogenosis tipo I (2-4 horas) y en la deficiencia de glucógeno sintetasa (4-8 horas). Puede ser algo más prolongado (10-20 horas) en las alteraciones de las enzimas gluconeogénicas y en la hipoglucemia cetósica idiopática. En los pacientes con alteraciones de la ?-oxidación y en aquellos con alteraciones de la cetogénesis y la cetólisis, la hipoglucemia suele aparecer tras ayunos muy prolongados (más de 16-24 horas). Se debe tener en cuenta que, en general, los niños pequeños son más vulnerables a la hipoglucemia de ayuno que los mayores.
• Las enfermedades metabólicas con frecuencia se manifiestan solo cuando el paciente se ve expuesto a situaciones estresantes que conllevan un aumento del catabolismo (infecciones, intervenciones quirúrgicas o traumatismos graves). Raramente, el ejercicio intenso puede desencadenar los síntomas, salvo en la excepcional forma de hiperinsulinismo, desencadenada por el ejercicio (mutaciones en SLC16A1: solute carrier family 16, member 1; AD).
• Es importante evaluar el ambiente sociofamiliar para detectar posibles casos de maltrato (hipoglucemia facticia secundaria a la administración de insulina o de hipoglucemiantes orales). La hipoglucemia secundaria a otras intoxicaciones suele ser reconocida en la anamnesis (etanol, sulfonilureas, salicilatos, quinina, etc.).
Exploración clínica
Algunos datos de la exploración pueden contribuir a orientar el diagnóstico:
• La presencia de talla baja, micropene o alteraciones de la línea media (paladar hendido, incisivo superior único, hipoplasia del nervio óptico…) deben hacer pensar en hipopituitarismos. En estos casos, la longitud al nacimiento es normal, pero el hipocrecimiento se hace evidente alrededor de los 5-6 meses de vida. Las enfermedades metabólicas, exceptuando las alteraciones de la β-oxidación de ácidos grasos, suelen cursar con escasa ganancia ponderal y de talla. El bajo peso se asocia también a la hipoglucemia cetósica idiopática. La obesidad o el antecedente de macrosomía neonatal es característica de los hiperinsulinismos.
• Deben buscarse rasgos sindrómicos, especialmente si existen antecedentes de macrosomía neonatal, por la posibilidad de hiperinsulinismos sindrómicos, como, por ejemplo: los síndromes de: Beckwith-Wiedemann (onfalocele, macroglosia, hemihipertrofia, surcos en el lóbulo auricular…), Kabuki o Silver-Rusell, entre otros.
• La presencia de genitales ambiguos y cariotipo 46, XX sugiere hiperplasia suparrenal congénita; mientras que, la hiperpigmentación cutánea pueden indicar la existencia de insuficiencia suprarrenal primaria (hipoplasia suprarrenal congénita, adrenalitis autoinmune…) o resistencia a la ACTH (por mutaciones en el receptor de ACTH o en su vía de señalización, o en el síndrome de Allgrove).
• Las enfermedades metabólicas suelen cursar con acidosis e hiperventilación y también es frecuente la afectación neurológica grave.
• La hepatomegalia puede ser muy evidente en las glucogenosis (la asociación de talla baja y hepatomegalia es característica). Una hepatomegalia moderada puede verse también en: las alteraciones de la gluconeogénesis, galactosemia, intolerancia a la fructosa, defectos de la β-oxidación de los AGL e, incluso, también, en los pacientes con hiperinsulinismo que reciben aportes elevados de glucosa para evitar la hipoglucemia. Si la hepatomegalia es dolorosa, se debe sospechar una enfermedad hepática aguda.
Pruebas complementarias básicas
La hipoglucemia espontánea supone el fracaso, por una u otra causa, de los mecanismos contrarreguladores. En situación de hipoglucemia, los niveles circulantes de determinados metabolitos y hormonas reflejan la integridad o la alteración de esos mecanismos contrarreguladores. Por ello:
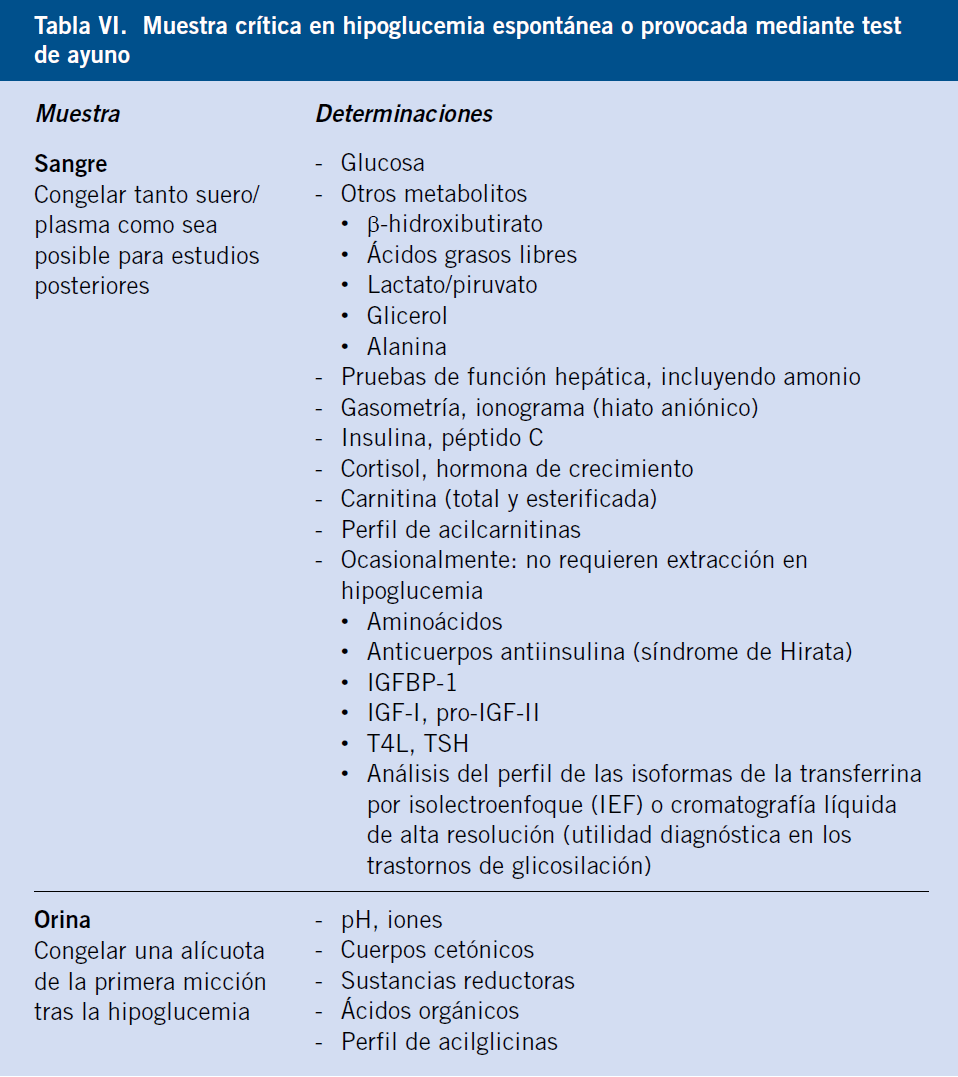
Los estudios analíticos (muestra crítica) para analizar la causa de una hipoglucemia deben efectuarse cuando los mecanismos de respuesta estén en marcha; es decir, en situación de hipoglucemia espontánea o bien en situación de hipoglucemia provocada, mediante test de ayuno. Estos estudios exigen disponer de una buena vía de acceso venosa.
• La muestra de sangre en hipoglucemia (muestra crítica) debe ser extraída, si es posible, sin utilizar compresor (aumento facticio de láctico y amonio), y antes de administrar ningún tratamiento. Dado que los parámetros a determinar son múltiples (Tabla VI), se recomienda disponer de una buena vía de acceso venosa que permita tanto las extracciones de sangre necesarias, como la administración posterior de medicación: glucagón (test de glucagón) o glucosa al 10% para revertir la hipoglucemia. La sangre debe ser enviada inmediatamente en hielo al laboratorio para iniciar el proceso diagnóstico. Debe recogerse también la orina de la primera micción tras el episodio de hipoglucemia.
• En caso de que no pueda extraerse sangre suficiente para realizar todas las determinaciones recomendadas, deberían cuantificarse los siguientes parámetros por este orden de prioridad: glucosa, β-hidroxibutirato, función hepática, AGL, insulina, láctico y cortisol. En Urgencias, como mínimo, GP y cetonemia/cetonuria. Es preferible cuantificar el β-hidroxibutirato en sangre, ya sea con métodos estándar de laboratorio o mediante un reflectómetro portátil, que la cetonuría; en cualquier caso, la orina debe ser analizada en busca de sustancias reductoras (Clinitest®) y de acetona (si β-hidroxibutirato en sangre no disponible). Cualquier excedente de plasma y la 1ª orina tras la hipoglucemia debería congelarse, preferiblemente a –70ºC, por si fuera necesario realizar determinaciones adicionales, posteriormente.
Test de tolerancia al ayuno y test de glucagón
Con bastante frecuencia, no es posible obtener las muestras críticas durante el primer episodio de hipoglucemia. En ese caso, si la causa de la hipoglucemia no es obvia (malnutrición, escasos aportes…), se debe ingresar al paciente y recurrir a la prueba de tolerancia al ayuno (test de ayuno):
• Siempre debe realizarse tras ingerir una dieta rica en carbohidratos durante, al menos, 3 días para saturar los depósitos hepáticos de glucógeno y evitar la aparición de una hipoglucemia facticia. La vía venosa de acceso debe estar bien establecida antes de plantearse el inicio de la prueba.
• Se trata de una prueba potencialmente peligrosa, por lo que siempre debe realizarse en régimen de hospitalización y retrasarse en caso de que el paciente presente cualquier enfermedad intercurrente, por leve que sea. Los riesgos son mayores en los pacientes con alteraciones del metabolismo de la carnitina o de la ?-oxidación de los AGL de cadena larga que, además de la hipoglucemia, pueden desarrollar arritmias (incluso parada cardíaca) durante la prueba, por lo que se debe excluir esta posibilidad antes de indicar una prueba de ayuno mediante la determinación del perfil de acilcarnitinas y la concentración de carnitina total y libre en sangre (puede hacerse con una muestra recogida en papel de filtro fuera del episodio de hipoglucemia). Se recomienda también excluir la posibilidad de miocardiopatía realizando un ecocardiograma previo.
• La duración máxima del ayuno debe ser decidida a priori en función de la edad del niño y del máximo tiempo que transcurre habitualmente entre dos tomas consecutivas. El momento de inicio de la prueba debe ser calculado según los datos anteriores para que la hipoglucemia aparezca preferiblemente durante las horas del día con más personal disponible, generalmente por las mañanas:
- En niños < 6 meses (rara vez es necesaria), la duración será de 8 h (inicio a las 4 a.m.).
- En niños de 6-8 meses, la duración será de 12 h (inicio a las 12 p.m.).
- En niños de 8-12 meses la duración será de 16 h (inicio a las 8 p.m.).
- En niños de 1-7 años la duración será de 18-20 h (inicio a las 4-6 p.m.).
- En niños > 7 años la duración será de 24 h (inicio a las 12 a.m.).
• Al comienzo de la prueba, habitualmente cuando se omite la primera toma (generalmente en torno a las 8 de la mañana), se realiza una gasometría, se determina la concentración de glucosa, β-hidroxibutirato, AGL, lactato, alanina e insulina, y se congela una alícuota de suero y orina. A partir de entonces, en función de la historia clínica y la edad del paciente, se determina la glucemia capilar periódicamente (cada 1-2 horas y si aparecen síntomas). Si durante la prueba, el paciente desarrolla hipoglucemia, se deben extraer las muestras críticas. En caso contrario, las muestras se deben extraer igualmente al final de la prueba. Es importante no olvidar la recogida de orina de la micción siguiente a la hipoglucemia. Algunos autores recomiendan, además, determinar la presencia de cetonuria en cada micción durante la prueba, o mejor β-OH-butírico capilar.
• Si se realiza adecuadamente, la prueba de ayuno es extremadamente útil en el diagnóstico de la hipoglucemia, hasta el punto de que raramente se necesita recurrir a otras exploraciones complementarias. Además, ofrece información útil para el tratamiento de los pacientes, ya que permite estimar el tiempo que el paciente puede permanecer en ayunas de forma segura.
• Si durante la prueba de ayuno el paciente desarrolla hipoglucemia, se debe realizar un test de glucagón después de extraer las muestras críticas (siempre y cuando la situación clínica del paciente permita retrasar el tratamiento con glucosa). Es especialmente útil en el diagnóstico diferencial de las hipoglucemias que se presentan en las primeras 8 horas de ayuno. Se realiza administrando 0,03 mg/kg de glucagón por vía i.m. o i.v. (máx.: 1 mg) y tomando muestras de sangre cada 10 minutos durante media hora. En los pacientes con hiperinsulinismo (endógeno o exógeno), se observa una marcada respuesta glucémica (elevación de glucemia >30-40 mg/dL); mientras que, una respuesta inferior a 20 mg/dL indica depleción de los depósitos de glucógeno (hipoglucemia cetósica idiopática) o incapacidad para convertir el glucógeno en glucosa (glucogenosis). Si a los 20’ de la administración de glucagón la elevación de la glucemia es inferior a 20 mg/dL, se debe suspender la prueba y administrar glucosa intravenosa para revertir la hipoglucemia.
Interpretación de los resultados
En condiciones normales, cuando la glucemia empieza a descender y se sitúa alrededor de 50 mg/dL: 1) los depósitos de glucógeno están exhaustos (no se produciría respuesta de glucemia a la administración de glucagón); 2) los niveles séricos de precursores neoglucogénicos han disminuido ligeramente (láctico < 1,5 mmol/L); 3) los AGL se han triplicado (1,5-2,0 mmol/L) y el β-hidroxibutírico, el principal cetoácido, se ha multiplicado por 50-100 (2-5 mmol/L); y 4) la insulinemia ha disminuido a valores prácticamente indetectables (< 2 µU/mL). La comparación de estos valores normales con los hallados en un paciente en situación de hipoglucemia, aporta la información básica para orientar el diagnóstico de la causa subyacente.
Una vez confirmada la hipoglucemia plasmática en el laboratorio (espontánea o inducida por el test de ayuno), el primer paso en el diagnóstico diferencial es analizar el grado de cetosis durante el episodio según los niveles plasmáticos de ?-hidroxibutirato (Fig. 2):

Figura 2. Algoritmo diagnóstico de la hipoglucemia en la infancia, fuera del periodo neonatal. AGL: ácidos grasos libres; EIM: error innato del metabolismo; GH: hormona de crecimiento; IGFs: factores de crecimiento semejantes a la insulina; T4L: tiroxina libre.
• Niveles elevados de β-hidroxibutirato (>2,5 mmol/L). Constituye el perfil endocrino-metabólico más habitual e inespecífico, conocido como hipoglucemia cetósica, e indica que el paciente es capaz de movilizar AGL desde el tejido adiposo y metabolizarlos a cuerpos cetónicos en el hígado. Se observa en la hipoglucemia cetósica idiopática, en algunas enfermedades metabólicas (glucogenosis, acidemias orgánicas) y en las deficiencias de hormonas contrarreguladoras después del período neonatal.
El diagnóstico diferencial dentro de este grupo requiere valorar, además de la historia clínica, la presencia de acidosis metabólica y la concentración de otros metabolitos y hormonas en el momento de la hipoglucemia. Durante la hipoglucemia, los niveles de hormonas contrarreguladoras (GH y cortisol) deben ser elevados (cortisol >20 µg/dL y GH >6 µg/L). Los valores inferiores deben ser considerados sospechosos y confirmados mediante la realización de las pruebas funcionales adecuadas (test de ACTH para cortisol y test de estimulación farmacológica de GH). La deficiencia de GH y/o cortisol durante el periodo neonatal y primer año de vida pueden presentarse solo con leve-moderada cetosis (<2,5 mmol/L).
Las concentraciones elevadas de lactato, piruvato, alanina y/o glicerol sugieren un defecto de la gluconeogénesis, de la glucogenolisis o de la glucolisis. La hiperlactacidemia sugiere una alteración de la gluconeogénesis (especialmente la deficiencia de fructosa-1,6-bisfosfatasa), de la glucogenolisis (glucogenosis tipo I) o de la β-oxidación de AGL de cadena larga. Las alteraciones de la cadena respiratoria mitocondrial y la deficiencia de piruvato carboxilasa también pueden producir hiperlactacidemia con hipoglucemia, pero generalmente se presentan de forma más crónica. Las glucogenosis tipo III y tipo VI raramente cursan con hiperlactacidemia. La cuantificación de ácidos orgánicos en orina es especialmente útil para diagnosticar las acidemias orgánicas.
• Cetosis leve o moderada (β-hidroxibutirato <2,5 mmol/L). La presencia de cetosis leve-moderada durante la hipoglucemia (hipoglucemia hipocetósica) indica que los AGL no están siendo movilizados adecuadamente desde los adipocitos (hiperinsulinismo) o no pueden ser metabolizados a cuerpos cetónicos (alteraciones de la ?-oxidación o de la cetogénesis).
La coexistencia de concentraciones muy bajas de β-hidroxibutirato (<0,5-1 mmol/L) y de AGL (<0,5 mmol/L) sugiere la existencia de hiperinsulinismo. Este diagnóstico se ve apoyado por unos niveles plasmáticos de insulina/péptido C elevados o inadecuadamente normales (si GP< 45 mg/dL: insulinemia < 2 µg/mL). Con mucha frecuencia, el diagnóstico de hiperinsulismo es difícil de comprobar debido a dos hechos fisiológicos fundamentales: 1) por un lado, al carácter pulsátil de la secreción de insulina y su corta vida media; y 2) por otro, a que se cuantifica la insulina en sangre venosa periférica y no en sangre portal, que es la que determina el efecto sobre la función metabólica del hígado y que puede ser 10 veces superior a la insulinemia periférica. En los casos dudosos, es necesario apoyarse en otros hallazgos para hacer el diagnóstico de hiperinsulinismo (Tabla V).
El hiperinsulinismo endógeno o secundario a intoxicación por sulfonilureas cursa con niveles elevados de péptido C; mientras que, niveles disminuidos de péptido C sugieren la administración de insulina exógena (insulinemia >100 µU/mL). La concentración medida de insulina en este último caso puede no ser elevada, si lo que se ha administrado son análogos de insulina, debido a la incapacidad de los anticuerpos monoclonales frente a la insulina humana para detectarlos.
La presencia de niveles elevados de AGL (>3 mmol/L) junto a una concentración baja de ?-hidroxibutirato sugiere una alteración de la ?-oxidación de los ácidos grasos o de la cetogénesis. En estos casos, la insulinemia suele estar adecuadamente suprimida (salvo en la deficiencia de hidroxiacil-coA-deshidrogenada por mutaciones en el gen SCHAD, que determina también hiperinsulinismo). Además de la hipoglucemia, los pacientes suelen presentar un mayor o menor grado de afectación multisistémica (debilidad muscular y miocardiopatía en la deficiencia primaria de carnitina o episodios de rabdomiolisis en la de carnitina-palmitoil-transferasa) con hiperuricemia y elevación de las transaminasas y la CPK, así como alteraciones neurológicas producidas por el efecto tóxico de algunos metabolitos (síndrome Reye-like). Debido a que muchos de los parámetros bioquímicos se normalizan fuera de las crisis, es importante recoger las muestras adecuadas durante la hipoglucemia y almacenarlas congeladas para su análisis posterior (excreción urinaria de ácidos dicarboxílicos, conjugados de glicina y carnitina; concentración plasmática de carnitina total y cociente acilcarnitinas/carnitina total).
Los pacientes con glucogenosis tipo I también pueden tener una cierta hipocetosis. Sin embargo, es relativamente sencillo distinguirlos de los anteriores por sus otras características clínicas y bioquímicas: gran hepatomegalia (salvo en el período neonatal), hipertrigliceridemia, ausencia de respuesta glucémica tras la administración de glucagón, pero marcado aumento de los niveles de lactato, etc.
Otras pruebas complementarias
En algunos casos, se puede recurrir a la realización de otras pruebas complementarias más específicas para confirmar el diagnóstico(30), como son: pruebas para evaluar la producción hepática de glucosa (sobrecargas i.v. de alanina, fructosa, galactosa…) o estudios funcionales que analizan la actividad de la enzima responsable en células del paciente (linfocitos, fibroblastos cutáneos, hígado, mucosa intestinal o músculo esquelético). Para el estudio de las hipoglucemias postprandiales, las pruebas de sobrecarga oral de glucosas o de alimento mixto pueden ser de utilidad. El desarrollo de la genética molecular ha relegado, en la mayoría de los casos, muchas de estas pruebas a un segundo plano.
Estudios moleculares
La confirmación diagnóstica definitiva de las hipoglucemias de origen genético requiere realizar el estudio de mutaciones en el correspondiente gen (HICP, glucogenosis, EIM…), generalmente en ADN obtenido de linfocitos de sangre periférica. En ocasiones, la gran mayoría de los pacientes tienen la misma mutación, lo que simplifica el estudio genético; por ejemplo, en el caso de la deficiencia de acil-CoA deshidrogenasa de cadena media (MCAD), la alteración más frecuente de la β-oxidación de los ácidos grasos (1:10.000 RN), en la que el 90% de los pacientes caucásicos presentan la mutación K329E (incluida en el cribado metabólico en países con alta frecuencia de esta mutación)(31). Otras veces, las mutaciones responsables de la enfermedad se distribuyen a lo largo del genoma, siendo necesario aplicar métodos de secuenciación masiva o next-generation sequencing (estudios de paneles de genes, exoma o genoma). En el caso de los HICP que no responden a diazóxido, se encuentra una causa genética en el 80-90% de los casos; mientras que, este porcentaje se reduce al 22-47% en los que sí responden a diazóxido(32). El hallazgo de mutaciones en ABCC8 y KCNJ11 en el alelo paterno puede sugerir hiperinsulinismo focal y posibilidad de curación completa mediante pancreatectomía parcial. En los pacientes con hiperinsulinismo congénito que se someten a pancreatectomía subtotal o parcial, puede ser conveniente, también, realizar estudios genéticos en el tejido pancreático extirpado. En los pacientes con insulinoma, debería realizarse sistemáticamente estudio de mutaciones asociadas a MEN1(24).
Pruebas de imagen
En los pacientes con hiperinsulinismo que no responden a diazóxido, antes de planificar la cirugía (pancreatectomía subtotal o parcial) es conveniente realizar un PET-TAC con 18-fluor-L-DOPA que puede permitir diferenciar entre formas focales y difusas de hiperinsulinismo(33,34). Esta técnica de reciente disponibilidad, requiere de personal experto para su interpretación, pero ha revolucionado el tratamiento quirúrgico de los hiperinsulinismos y relegado a otras técnicas de imagen (ecografía de alta resolución, TAC-RM abdominal o arteriografía selectiva del tronco celíaco) o pruebas de localización técnicamente difíciles de realizar y que no suelen estar disponibles (cateterización venosa selectiva por vía percutánea transhepática). La sensibilidad y especificidad medias del PET-TAC con 18-fluor-L-DOPA para distinguir entre hiperinsulinismo focal y difuso, en un reciente metanálisis(35), fue del 89% (entre 81-95%) y 98% (entre 89 y 100%), respectivamente. En el caso de los adenomas, la prueba de imagen de primera línea es la RM(24), con sensibilidades que oscilan entre el 30 y el 85%, debido, probablemente, a que el tamaño tumoral puede ser, en ocasiones, muy pequeño; en estos casos, el PET-TAC con 18-fluor-L-DOPA podría estar indicado.
En pacientes con deficiencias hormonales, puede ser necesario realizar una RM hipotálamo-hipofisaria y/o estudios de imagen de las glándulas suprarrenales.
Orientación terapéutica
Tratamiento urgente
El tratamiento agudo de la hipoglucemia se basa en aportar al paciente la glucosa necesaria para normalizar la glucemia, evitando siempre la sobrecorrección. En EIM con acidosis metabólica suele estar indicada, también, la administración de bicarbonato.
En los casos graves o cuando existe un acceso vascular disponible
Se debe administrar un bolo i.v. de 200 mg/kg de glucosa en forma de suero glucosado al 10% (2 mL/kg). No deben infundirse soluciones más concentradas por vía periférica, dado el alto riesgo de lesión tisular en caso de extravasación. El bolo debe administrarse lentamente (2-3 mL/min.) para evitar la hiperglucemia, que podría dar lugar a la liberación de insulina y a una hipoglucemia reactiva. Tras el bolo, se iniciará una perfusión continua de glucosa a un ritmo de 6-8 mg/kg/minuto en lactantes y 3-5 mg/kg/minuto en el niño mayor. El ritmo debe ser continuamente ajustado en función de los sucesivos controles de glucemia para mantenerla por encima de 70 mg/dL (>100 mg/dl ante la sospecha de EIM). Los controles deben realizarse inicialmente, al menos, cada 30-60 minutos, hasta que la glucemia sea estable y cada 2-4 horas después. Nunca debe interrumpirse bruscamente la administración de glucosa por el riesgo de hipoglucemia de rebote. El ritmo de la infusión de glucosa se descenderá progresivamente a medida que el niño tolere la alimentación oral o responda al tratamiento farmacológico o dietético específico.
Los requerimientos de glucosa, para mantener glucemias >70 mg/dL, superiores a 8-10 mg/kg/min sugieren la existencia de un hiperinsulinismo, pudiendo llegar, en ocasiones, a los 20-25 mg/kg/min. En estos casos, para minimizar la sobrecarga de volumen, puede recurrirse a la administración de soluciones de glucosa hipertónica a través de una vía central, así como asociar temporalmente una perfusión continua de glucagón i.v. o s.c. (5-20 µg/kg/hora) y/o octreótido s.c. (5-40 µg/kg/día). La hipoglucemia secundaria a sobredosis de sulfonilureas se trata con bolos de glucosa i.v. y, en caso de que no sea suficiente, con octreótido i.m. o s.c. (1-1,5 µg/kg; máximo: 150 µg, cada 6 horas).
En hipoglucemias graves sin acceso venoso
Se pueden tratar con glucagón i.m. o s.c. (0,03 mg/kg/dosis; dosis máxima: 1 mg). El glucagón es efectivo, sobre todo, en los pacientes diabéticos o con hiperinsulinismo, que poseen grandes depósitos de glucógeno susceptible de ser liberado a la sangre, pero la respuesta suele ser transitoria (alrededor de una hora); por lo que, puede ser necesario administrar más de una dosis si la tendencia a la hipoglucemia persiste.
Medidas terapéuticas diferidas
El tratamiento específico a largo plazo depende de la causa de la persistencia de la hipoglucemia y, en gran medida, se ha comentado a lo largo del artículo. La guía de la Sociedad Americana de Endocrinología Pediátrica(7), recomienda como objetivos glucémicos:
• Para RN con sospecha de hipoglucemia persistente y para el resto de los niños con trastorno hipoglucémico confirmado, mantener GP > 70 mg/dL.
• Para RN con alto riesgo de hipoglucemia, pero sin sospecha de trastorno hipoglucémico congénito, mantener la GP > 50 mg/dL, en menores de 48 horas, y > 60 mg/dL en mayores de 48 horas.
• Se recomienda un enfoque individualizado del tratamiento en función de la enfermedad responsable de la hipoglucemia, teniendo en consideración la seguridad del paciente y las preferencias de la familia.
Medidas dietéticas
Los pacientes con hipoglucemia cetósica idiopática, glucogenosis, alteraciones de la gluconeogénesis, de la β-oxidación de los ácidos grasos e hiperinsulinismos leves deben evitar los ayunos prolongados, realizando tomas más o menos frecuentes en función de la enfermedad de base con una dieta personalizada; aunque, en todos los casos, se recomiendan preferentemente carbohidratos de absorción lenta. Los pacientes con glucogenosis tipo I pueden requerir la administración continua, mediante una sonda nasogástrica o gastrostomía, de polímeros de glucosa o maltodextrina (almidón de maíz crudo en niños mayores y adultos). La dieta de los pacientes con alteraciones de la ?-oxidación o de la cetogénesis debe ser pobre en grasas, aunque en caso de alteración comprobada de la β-oxidación de AGL de cadena larga, puede aumentarse el contenido calórico de la dieta añadiendo MCT (triglicéridos de cadena media). Los pacientes con intolerancia hereditaria a la fructosa deben evitar la ingesta de fructosa (azúcar de mesa, frutas, miel, muchos jarabes) y los pacientes con galactosemia, los alimentos y fármacos que contengan galactosa o lactosa.
Algunos de los EIM, así como la hipoglucemia hipocetósica idiopática, pueden descompensarse durante las enfermedades intercurrentes. En estas situaciones, deben ofrecerse frecuentemente líquidos con alto contenido en hidratos de carbono y, si la ingesta resulta inadecuada, administrar glucosa por vía i.v. No deben administrarse lípidos i.v. a pacientes con alteraciones de la β-oxidación o de la cetogénesis.
Hiperinsulinismo(20-21,36-39)
En las formas más leves, puede ser suficiente con realizar tomas frecuentes de alimentos con bajo índice glucémico. En los demás casos, el fármaco de primera elección es el diazóxido v.o. (5-15 mg/kg/día en 3 dosis), una droga que actúa abriendo los canales de K dependientes de ATP. El diazóxido produce hirsutismo y retención hídrica, especialmente en el periodo neonatal; en este caso, puede asociarse hidroclorotiazida v.o. (7 mg/kg/día en 2 dosis; máxima dosis: 10 mg/kg/día). Más raramente aparecen otros efectos adversos graves que obligan a interrumpir el tratamiento (neutropenia, trombopenia, insuficiencia cardíaca). Se considera que no hay respuesta a diazóxido cuando: después de, al menos, 5 días de tratamiento con diazóxido a dosis de 15 mg/kg/día, el niño sigue requiriendo, para mantener su glucemia normal, aportes intravenosos de glucosa y/o es incapaz de mantener la GP durante una noche de ayuno. La respuesta clínica a diazóxido marca un antes y un después, en la orientación diagnóstico-terapéutica de los pacientes con HICP (Fig. 3).

Figura 3. Algoritmo diagnóstico de la hipoglucemia persistente por hiperinsulinismo. Modificado de referencias 20 y 21.
Si no hay respuesta a diazóxido, puede considerarse la asociación de análogos de somatostatina, como: octreótido s.c. (5-25 µg/kg/día c/6-8 h, máximo: 40 µg/kg/día) o lanreótido (Somatulina Autogel®: 30-60 mg/mes, s.c.). La administración continuada de análogos de somatostatina, se ha asociado a enterocolitis necrotizante, en el periodo neonatal, y, en edades posteriores, puede producir taquifilaxia, esteatorrea, colelitiasis y alteración del crecimiento. Recientemente, se han incorporado nuevos fármacos, todavía insuficientemente probados, al tratamiento del hiperinsulinismo, como son: exendin, un antagonista del receptor de GLP1 (glucagón-like peptide 1), y sirolimus (rapamicina), un inmunomodulador, que inhibe la vía mTOR (mammalian target of rapamycin) y genera insulinorresistencia por un mecanismo insuficientemente aclarado, pero que ha sido utilizado en el tratamiento de insulinomas malignos.
Si el tratamiento farmacológico no es eficaz, el PET-TAC con 18-fluor-L-dopa muestra un hiperinsulinismo focal o atípico o se sospecha un adenoma productor de insulina, debe transferirse el paciente a un centro especializado para realizar una pancreatectomía, preferiblemente por vía laparoscópica(38). La pancreatectomía será más o menos extensa, según los hallazgos del PET-TAC con 18-fluor-L-DOPA. En caso de hiperinsulinismos difusos que no responden a la terapia médica, pueden ser necesarias pancreatectomías casi-totales (95-98%).
Evolución
El pronóstico de los pacientes con hipoglucemia depende de la enfermedad subyacente. La hipoglucemia cetósica idiopática tiende a resolverse espontáneamente en torno a los 5 años, cuando el niño adquiere la masa muscular y el tejido adiposo suficientes para prevenir la hipoglucemia en caso de ayuno. Las enfermedades metabólicas y las deficiencias hormonales requieren tratamiento de por vida, que puede, en ocasiones, ser muy efectivo para prevenir la recurrencia de los episodios de hipoglucemia, pero no tanto para el pronóstico general, determinado por la enfermedad de base. En los pacientes con hiperinsulinismo, el pronóstico depende fundamentalmente de la causa del mismo. El hiperinsulinismo leve que responde a diazóxido puede necesitar tratamiento continuado, pero permite realizar una vida normal; en algunos casos, se puede producir una remisión espontánea y dejar de requerir tratamiento. Se puede probar a retirar el tratamiento cuando se requieran < 5 mg/kg/día de diazóxido o < 5 µg/kg/día de octreótido para mantener glucemias por encima de 70 mg/dl y se tolere un test de ayuno, adecuado a su edad y peso. Las lesiones focales se curan con pancreatectomía parcial. En las formas difusas que no responden al tratamiento farmacológico, casi exclusivas del período neonatal, la pancreatectomía subtotal puede no controlar completamente las hipoglucemias (con secuelas neurocognitivas a largo plazo frecuentes), requiriendo medicación (diazóxido, octerótido…) o, por el contrario, producir diabetes mellitus secundaria e insuficiencia pancreática exocrina.
Papel del pediatra de Atención Primaria (AP)
Aunque el estudio de las hipoglucemias persistentes o recurrentes es una patología de ámbito hospitalario, el papel del pediatra de AP en estas patologías es muy importante, sobre todo en el periodo de lactante, donde la inespecificidad de las hipoglucemias obliga a este a mantener un alto índice de sospecha que haga posible un diagnóstico más precoz que pueda disminuir/evitar las secuelas neurológicas que, de mayor o menor entidad, se asocian a las hipoglucemias reiteradas. Además, la hipoglucemia cetósica idiopática, la forma de hipoglucemia más frecuente entre los 18 meses y los 5 años de edad es patrimonio del pediatra de AP. Esta entidad es fácilmente diagnosticada sin necesidad de costosos estudios complementarios, por la edad de inicio, los antecedentes personales (bajo peso al nacimiento, malos comedores…) y el contexto clínico en que se desarrollan los episodios de hipoglucemia, habitualmente leves. Estos pacientes pueden ser perfectamente controlados mediante consejos de alimentación y prevenidas las hipoglucemias mediante la determinación de cetonuria con tiras reactivas, en su domicilio, durante los procesos infecciosos intercurrentes, a la espera de su desaparición espontánea con la edad; no obstante, en caso de hipoglucemias graves o reiteradas, deberán ser remitidos para su estudio hospitalario.
Bibliografía
Los asteriscos reflejan el interés del artículo a juicio del autor.
1. Rubio O, Pozo J. Hipoglucemia en el niño. En: Casado J, Serrano A, eds. Urgencias pediátricas graves. Síntomas guía, técnicas y cuidados intensivos (3ª edición). Madrid: Ergon S.A., 2015; p. 1375-82.
2. De León, DD, ThorntonPS, Stanley CA, Sperling MA. Hypoglycemia in the newborn and infant. En: Sperling MA, ed. Pediatric Endocrinology (4th edition). Philadelphia: Elsevier Saunders, 2014; p. 157-85.
3. Flykanaka-Gantenbein C. Hypoglycemia in childhood: long term effects. Pediatr Endocrinol Rev. 2004; 1 (Suppl. 3): 530-36.
4. Shah R, Harding J, Brown J, McKinlay C. Neonatal Glycaemia and Neurodevelopmental Outcomes: A Systematic Review and Meta-Analysis. Neonatology. 2018; 115: 116-26.
5. Cryer PE. Mechanisms of hypoglycemia-associated autonomic failure in diabetes. N Engl J Med. 2013; 369: 362-72.
6. Cryer PE. Hypoglycemia. En: Melmed S, Polonsky KS, Larsen PR, Kronnenberg HM, eds. Williams Textbook of Endocrinology (12th edition). Philadelphia: Elsevier Saunders, 2011; p. 1552-80.
7.** Thornton PS, Stanley CA, De Leon DD, Harris D, Haymond MW, Hussain K, et al. Recommendations from the Pediatric Endocrine Society for Evaluation and Management of Persistent Hypoglycemia in Neonates, Infants, and Children. J Pediatr. 2015; 167: 238-45.
8. Moore MC, Connolly CC, Cherrington AD. Autoregulation of hepatic glucose production. Eur J Endocrinol. 1998; 138: 240-8.
9. Langdon DR, Stanley CA, Sperling MA. Hypoglycemia in the toddler and child. En: Sperling MA, ed. Pediatric Endocrinology (4th edition). Philadelphia: Elsevier Saunders, 2014; p. 920-55.
10. Stanley CA, Rozance PJ, Thornton PS, De Leon DD, Harris D, Haymond MW, et al. Re-evaluating “transitional neonatal hypoglycemia”: mechanism and implications for management. J Pediatr. 2015; 166: 1520-5.e1.
11. Lang TF, Hussain K. Pediatric hypoglycemia. Adv Clin Chem. 2014; 63: 211-45.
12. Güemes M, Rahman SA, Hussain K. What is a normal blood glucose? Arch Dis Child. 2016; 101: 569-574.
13. Abraham MB, Jones TW, Naranjo D, Karges B, Oduwole A, Tauschmann M, Maahs DM. ISPAD Clinical Practice Consensus Guidelines 2018: Assessment and management of hypoglycemia in children and adolescents with diabetes. Pediatr Diabetes. 2018; 19 (suppl.27): 178-192. Disponible en: https://doi.org/10.1111/pedi.12698.
14. Cryer PE, Axelrod L, Grossman AB, Heller SR, Montori VM, Seaquist ER, et al. Endocrine Society. Evaluation and management of adult hypoglycemic disorders: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2009; 94: 709-28.
15. Boluyt N, van Kempen A, Offringa M. Neurodevelopment after neonatal hypoglycemia: a systematic review and design of an optimal future study. Pediatrics. 2006; 117: 2231-43.
16. Cornblath M, Hawdon JM, Williams AF, Aynsley-Green A, Ward-Platt MP, Schwartz R, et al. Controversies regarding definition of neonatal hypoglycemia: suggested operational thresholds. Pediatrics. 2000; 105: 1141-5.
17. Tin W. Defining neonatal hypoglycaemia: a continuing debate. Semin Fetal Neonatal Med. 2014; 19: 27-32.
18. Adamkin DH and Committee on fetus and newborn. Postnatal Glucose Homeostasis in Late-Preterm and Term Infants. Pediatrics. 2011; 127: 575-79.
19. Martín I, Güemes M, Guerrero-Fernández J, Salamanca L. Hipoglucemia. Manejo Diagnóstico-Terapéutico inicial. En: Guerrero-Fernández J, González-Casado I eds. Manual de diagnóstico y terapéutica en Endocrinología Pediátrica. Madrid: Ergon, 2018; p. 205-20.
20.** Güemes M, Hussain K. Hyperinsulinemic hypoglycemia. Pediatr Clin N Am. 2015; 62: 1017-36.
21.** Galcheva S, Al-Khawaga S, Hussain K. Diagnosis and management of hyperinsulinaemic hypoglycaemia. Best Pract Res Clin Endocrinol Metab. 2018; 32: 551-73. doi: 10.1016/j.beem.2018.05.014.
22. Stanley CA. Perspective on the genetics and diagnosis of congenital hyperinsulinism disorders. J Clin Endocrinol Metab. 2016; 101: 815-26.
23. Treberg JR, Clow KA, Greene KA, Brosnan ME, Brosnan JT. Systemic activation of glutamate dehydrogenase increases renal ammoniagenesis: implications for the hyperinsulinism/hyperammonemia syndrome. Am J Physiol Endocrinol Metab. 2010; 298: E1219-25.
24. Padidela R, Fiest M, Arya V, Smith VV, Ashworth M, Rampling D, et al. Insulinoma in childhood: clinical, radiological, molecular and histological aspects of nine patients. Eur J Endocrinol. 2014; 170: 741-7.
25. Hirata Y, Uchigata Y. Insulin autoimmune syndrome in Japan. Diabetes Res Clin Pract. 1994; 24 (Suppl): S153-S157.
26. Güemes M, Melikyan M, Senniappan S, Hussain K. Idiopathic postprandial hyperinsulinaemic hypoglycaemia. J Pediatr Endocrinol Metab. 2016; 29: 915-22. DOI 10.1515/jpem-2016-0043.
27. Sanjurjo P, Baldellou A. Diagnóstico y tratamiento de las enfermedades metabólicas hereditarias (3ª edición). Madrid: Ergon S.A. 2010.
28. Gil Ortega. Protocolos de diagnóstico y tratamiento de los errores congénitos del metabolismo (2ª edición) (AECOM). Madrid: Ergon 2018.
29. Martínez-Pardo M, Gómez L, Ruiz M, Sánchez-Valverde F, Dalmau J. Diagnóstico y tratamiento de las alteraciones del metabolismo de los carbohidratos. En: Protocolos diagnóstico-terapéuticos de Gastroenterología, Hepatología y Nutrición Pediátrica (SEGHNP-AEP). Madrid: Ergon 2010; p. 371-8.
30. Rubio O, Argente J. Metodología diagnóstica de la hipoglucemia en la infancia. Hormona y Factores de Crecimiento. 2008; XI: 9-22.
31. Matsubara Y, Narisawa K, Tada K. Medium-chain acyl-CoA dehydrogenase deficiency: molecular aspects. Eur J Pediatr. 1992; 151(3): 154-9.
32. Kapoor RR, Flanagan SE, Arya VB, Shield JP, Ellard S, Hussain K. Clinical and molecular characterisation of 300 patients with congenital hyperinsulinism. Eur J Endocrinol. 2013; 168: 557-64.
33. Christiansen CD, Petersen H, Nielsen AL, Detlefsen S, Brusgaard K, Rasmussen L, et al. 18F-DOPA PET/CT and 68Ga-DOTANOC PET/CT scans as diagnostic tools in focal congenital hyperinsulinism: a blinded evaluation. Eur J Nucl Med Mol Imaging. 2018; 45: 250-61. doi: 10.1007/s00259-017-3867-1.
34. Arbizu J, Sancho L. PET/CT con 18-FDopa en la diferenciación de las formas focales y difusas de hiperinsulinismo. Rev Esp Endocrinol Pediatr. 2015; 6 (Suppl): 27-31.
35. Treglia G, Mirk P, Giordano A, Rufini V. Diagnostic performance of fluorine-18-dihydroxyphenylalanine positron emission tomography in diagnosing and localizing the focal form of congenital hyperinsulinism: a meta-analysis. Pediatr Radiol. 2012; 42: 1372-9.
36. Kapoor RR, Flanagan SE, Arya VB, Shield JP, Ellard S, Hussain K. Clinical and molecular characterisation of 300 patients with congenital hyperinsulinism. Eur J Endocrinol. 2013; 168: 557-64.
37. Yorifuji T. Congenital hyperinsulinism: current status and future perspectives. Ann Pediatr Endocrinol Metab. 2014; 19: 57-68.
38. Maiorana A, Dionisi-Vici C. Hyperinsulinemic hypoglycemia: clinical, molecular and therapeutical novelties. J Inhert Metab Dis. 2017; 40: 531-42. doi: 10.1007/s10545-017-0059-x.
39. Martínez L, Chocarro G. Tratamiento quirúrgico de las formas focales de hiperinsulinismo. Rev Esp Endocrinol Pediatr. 2015; 6 (Suppl): 33-7.
Bibliografía recomendada:
– Thornton PS, Stanley CA, De Leon DD, Harris D, Haymond MW, Hussain K, et al. Recommendations from the Pediatric Endocrine Society for Evaluation and Management of Persistent Hypoglycemia in Neonates, Infants, and Children. J Pediatr. 2015; 167: 238-45.
Excelente artículo con las recomendaciones de la Sociedad Americana de Endocrinología Pediátrica respecto a cuándo y cómo deben ser estudiadas las hipoglucemias en la infancia, para evitar un excesivo gasto en pruebas innecesarias.
– Güemes M, Hussain K. Hyperinsulinemic hypoglycemia. Pediatr Clin N Am. 2015; 62: 1017-36.
– Galcheva S, Al-Khawaga S, Hussain K. Diagnosis and management of hyperinsulinaemic hypoglycaemia. Best Pract Res Clin Endocrinol Metab. 2018; 32: 551-73. doi: 10.1016/j.beem.2018.05.014.
Dos excelentes revisiones, del mismo grupo de trabajo, que ponen al día las hipoglucemias por hiperinsulinismo, la causa más frecuente de hipoglucemias persistentes en el periodo neonatal y uno de los campos que más ha evolucionado en los últimos años, desde el punto de vista diagnóstico (valor de la 18-fluor-L-dopa), molecular (nuevos y viejos genes implicados) y terapéutico (descripción de formas histológicas focales, susceptibles de curación mediante cirugía, y el empleo experimental de nuevos fármacos).
| Caso clínico |
|
Niño de 16 meses que fue remitido para estudio por hipoglucemia. Refería en los últimos 4 meses, tres episodios de convulsión tónico-clónica generalizada, sin fiebre ni aparente causa desencadenante, en la última de las cuales se había detectado una glucemia capilar de 27 mg/dL. Los padres no referían otra sintomatología sugerente de hipoglucemia. Ambos progenitores eran jóvenes y sanos, no consanguíneos, sin antecedentes de enfermedades familiares, ni de abortos o mortinatos y el paciente tenía un hermano de 4 años sano. Había nacido tras un embarazo normal y un parto a término, eutócico, con peso y longitud al nacimiento de 3.500 g y 50 cm, respectivamente. Desarrollo psicomotor normal y sin otros antecedentes personales de interés. La exploración física era normal, con genitales normales (estadio I de Tanner), peso de 10,9 kg (PC 75-90) y longitud de 81,5 cm (PC 25-50). El paciente fue ingresado para estudio, realizándose un perfil glucémico durante 48 horas que confirmó la tendencia a presentar glucemias en el rango bajo de la normalidad (60-70 mg/dL) con hipoglucemias leves esporádicas (40-45 mg/dL), preferentemente postprandiales y con escasa sintomatología acompañante (decaimiento, palidez). El control de la glucemia precisó inicialmente aportes de glucosa intravenosa de hasta 8 mg/kg/min. Tras su estabilización, se inició alimentación oral, con dieta fraccionada cada 4 horas, junto con nutrición enteral nocturna a débito continuo, pese a lo cual continuó presentando episodios de hipoglucemia, por lo que se instauró nutrición enteral continua con aportes elevados de hidratos de carbono, normalizándose las cifras de glucemia. Las determinaciones analíticas realizadas en hipoglucemia espontánea (muestra crítica), en sangre (insulina, péptido C, amonio, ácido láctico y pirúvico, hormona de crecimiento, cortisol, cuerpos cetónicos, ácidos grasos libres, alanina, aminoácidos, carnitina y acilcarnitina) y orina (cuerpos cetónicos y ácidos orgánicos en orina de 24 horas) fueron normales, salvo la presencia de niveles detectables de insulina (16,9 µUI/mL) e hiperamoniemia (147 µmol/L; VN: 10-40) en presencia de hipoglucemia (glucemia plasmática de 38 mg/dL). El paciente fue diagnosticado de hiperinsulinismo pautándose tratamiento con diazóxido oral (13,6 mg/kg/día, c/8 horas). Se observó respuesta clínica positiva a las 48 horas de iniciado el tratamiento, lo que permitió la retirada progresiva de los aportes endovenosos de glucosa, así como de la alimentación enteral y el paso a la alimentación fraccionada, con posterior descanso nocturno. El estudio molecular demostró que el paciente era heterocigoto para una mutación de novo en el exón 7 del gen GLUD-1 (G979A), confirmando el diagnóstico de síndrome de hiperinsulinimo hiperamoniemia.
|
