 |
| El Rincón del Residente |
Coordinadores:
M. García Boyano*, I. Noriega Echevarría**, E. Pérez Costa*, D. Rodríguez Álvarez*
*Residentes de Pediatría del Hospital Universitario Infantil La Paz. Madrid. **Residente de Pediatría del Hospital Universitario Infantil Niño Jesús. Madrid
Autores:
M. García Gozalo*, M. Furones García*, E. Lara Orejas**
*Residente de Pediatría. Hospital Universitario Infanta Cristina.**Adjunta de Pediatría. Servicio de Endocrinología. Hospital Universitario Infanta Cristina
|
El Rincón del Residente es una apuesta arriesgada de Pediatría Integral. No hemos querido hacer una sección por residentes para residentes. Yendo más allá, hemos querido hacer una sección por residentes para todo aquel que pueda estar interesado. Tiene la intención de ser un espacio para publicaciones hechas por residentes sobre casos clínicos e imágenes entre otras. |
Pediatr Integral 2019; XXIII (4): 223.e1 – 223.e6
Imagen en Pediatría Clínica.
Haz tu diagnóstico
Genitales ambiguos en recién nacido
Historia clínica
Recién nacido a término, con embarazo controlado de curso normal. Edad gestacional (EG) de 40 semanas. Apgar 9/10. Al nacimiento, presenta hipoglucemia precoz asintomática resuelta. Antropología al nacimiento: peso: 4.480 g (p>90; +3,2 SDS para EG), longitud: 54 cm (p>90; +2,5 SDS para EG) y perímetro cefálico: 34 cm (P25-50; -0,37 SDS para EG). La exploración física es normal salvo los genitales, que presentan hipertrofia de clítoris, meato uretral en base y labios mayores que sugieren escroto hipoplásico, en los que no se palpan masas (estadio II de Prader) (Figs. 1 y 2).
• Pruebas complementarias:
- Analítica a los 3 días de vida: hemograma y bioquímica normales, con sodio de 144 mmol/L y potasio de 5,1 mmol/L; testosterona >15 ng/ml (1.7-3.8); 17-0H-progesterona: 417,6 ng/ml (3,30 – 10,94) ; Cortisol: 9 mcg/dl (1.95-29.51) ; DHEAS <15 mcg/ml (6 – 160); Androstenediona: 4,4 ng/ml (0,9 – 5.3) ; y ACTH: 653 pg/ml (9.4-158.4).
- Ecografía abdominal: llama la atención que, a pesar de no existir una clara hipertrofia cortical, ambas suprarrenales están aumentadas de tamaño, sin presencia de nódulos ni masas. No se logra identificar ovarios y no se identifican imágenes sugestivas de testículos en cavidad abdominal. Se observa cavidad uterina con ratio cuerpo/cérvix normal para la edad.
- Cariotipo 46, XX.
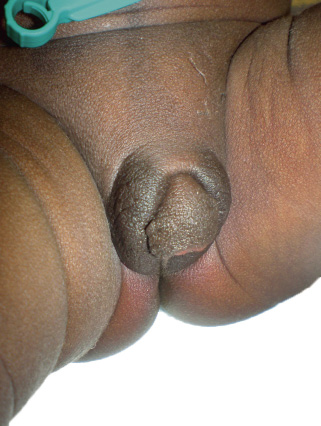
Figura 1.
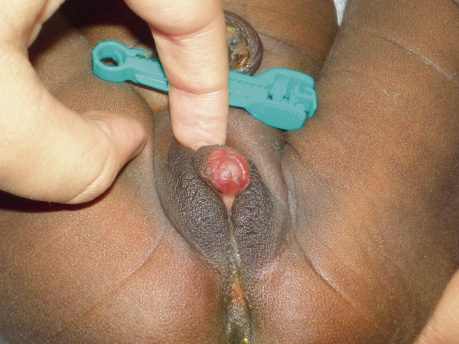
Figura 2.
¿Cuál es el diagnóstico?
a. Deficiencia de aromatasa placentaria.
b. Disgenesia gonadal.
c. Deficiencia de 5 alfa reductasa.
d. Hijo de madre con hiperplasia suprarrenal congénita (HSC) no tratada.
e. HSC por deficiencia de 21 hidroxilasa.
Respuesta correcta e. HSC por deficiencia de 21 hidroxilasa.
Ver comentario
Comentarios
a. La deficiencia de aromatasa placentaria resulta de una acumulación de andrógenos placentarios y una producción deficiente de estrógenos, que puede virilizar tanto a la madre como al feto 46, XX(1). En este caso, el producto en exceso serán los andrógenos adrenales que no se metabolizan a estrógenos por falta de aromatasa placentaria. En este caso no existiría una elevación tan marcada de la 17-OH progesterona.
b. En la disgenesia gonadal en niñas 46, XX, el defecto en la producción de hormonas femeninas no altera el desarrollo genital, por lo tanto, no habrá genitales ambiguos, sino un fenotipo femenino normal con hipogonadismo hipergonadotropo que se traducira en la pubertad con un retraso/ausencia de inicio del desarrollo puberal y amenorrea.
c. La deficiencia de 5 alfa reductasa impide la conversión de testosterona en dihidrotestosterona (DHT)(2), responsable de la virilización de los genitales externos. En los recién nacidos con cariotipo 46, XY, se pondrá de manifiesto al nacimiento con una ambigüedad genital por virilización variable, pero incompleta; por el contrario, en los RN con cariotipo 46, XX, el fenotipo será femenino normal.
d. En la hiperplasia suprarrenal materna no tratada en raras ocasiones puede provocar una virilización en el feto femenino salvo que sufra la misma enfermedad. Además las mujeres con HSC clásicas no tratada suelen ser infértiles.
e. La hiperplasia suprarrenal congénita es un grupo de desórdenes de herencia autosómica recesiva que se producen por una alteración a nivel de la esteroidogénesis suprarrenal. Existen cuatro deficiencias enzimáticas que provocan virilización del feto femenino 46, XX. La más frecuente es la mutación en el gen de la enzima 21 hidroxilasa. Esta enzima es responsable de la conversión de la 17-hidroxiprogesterona (17-OHP) en 11-deoxicortisol (precursor del cortisol) y de la progesterona en deoxicosticosterona (precursor de la aldosterona).
Discusión
Existen dos formas de HSC por deficiencia de 21-hidroxilasa: las formas clásicas con/sin pérdida salina, formas virilizante pura y pierde sal, respectivamente; y las formas no clásicas, en las que no se produce virilización de genitales externos (a veces, mínima clitoromegalia) y que suelen manifestarse en la mayoría de los casos, en el periodo prepuberal, por adrenarqua prematura (pubarquia, axilarquia, cambio en el olor corporal...), talla alta y maduración ósea acelerada. En la pubertad, las formas no clásicas pueden ser responsables de un cierto grado de hiperandrogenismo y pueden asociarse a síndrome de ovario poliquístico. Las formas clásicas de deficiencia de 21-hidroxilasa se diagnostican habitualmente en el cribado neonatal (prueba del talón); por el contrario, las formas no clásicas escapan a dicho cribado y pueden precisar para su diagnóstico bioquímico la realización de un test de ACTH para 17-OHP (pico de 17-OHP ≥ 10 ng/mL a los 60’). El diagnóstico definitivo de ambas formas de HSC se establece mediante el estudio molecular del gen de la 21-hidroxilasa, imprescindible para un adecuado consejo genético.
En el periodo neonatal, en el recién nacido a término, se consideran normales valores de 17-OHP <35 ng/ml en suero(3). Por encima de esta cifra, puede sospecharse deficiencia de 21-hidroxilasa. Esta entidad se acompaña de elevación de andrógenos y del tamaño de la glándula suprarrenal. Una vez diagnosticado, es fundamental descartar la pérdida salina, que se manifiesta clínicamente alrededor de la segunda semana de vida y que puede comprometer la vida del recién nacido.
El tratamiento de estos pacientes consiste en hidrocortisona a una dosis que minimice el máximo posible los efectos adversos. Así mismo, se iniciará 9-fludrocortisona en formas clásicas (causa más frecuente de ambigüedad sexual).
Palabras clave
Genitales ambiguos; Trastorno del desarrollo sexual; Hiperplasia suprarrenal congénita; Disgenesia gonadal; Ambiguous genitalia; Disorders of sex development; Congenital adrenal hiperplasia; Gonadal dysgenesis.
Bibliografía
1. Audí L, et al. Anomalías de la diferenciación sexual. An Pediatr Contin. 2011; 9(1): 15-30.
2. Murphy C, et al. Ambiguous Genitalia in the Newborn: An Overview and Teaching Tool. Journal of Pediatric and Adolescent Gynecology. 2011; 24: 236-50.
3. Rodríguez Sánchez A, et al. Hiperplasia suprarrenal congénita por déficit de 21-hidroxilasa. Pediatría Integral. 2015; XIX(7): 488-97.
