 |
| Regreso a las bases |
J.C. López Robledillo
Unidad de Reumatología Pediátrica. Hospital Infantil Universitario Niño Jesús Madrid
Pediatr Integral 2017; XXI (3): 219.e1 – 219.e16
Anamnesis y exploración física en Reumatología
Introducción
Las enfermedades reumáticas (ER) en niños y adolescentes, constituyen un grupo importante de procesos que afectan al sistema musculoesquelético de forma predominante, pero que, con frecuencia, comprometen también a otros órganos o sistemas y cuyos síntomas y signos pueden orientarnos a su diagnóstico. El espectro de las ER es amplio y abarca procesos locales y transitorios de índole mecánica, dolor crónico musculoesquelético, osteocondrosis, infecciones musculoesqueléticas, fragilidad ósea, enfermedad inflamatoria crónica articular y enfermedades sistémicas autoinmunes o autoinflamatorias.
Las ER en niños y adolescentes son, por lo general, procesos crónicos que se presentan en cualquier grupo etario y que podemos clasificar de forma operativa según la tabla I.

Por otro lado, es importante tener en cuenta que determinadas enfermedades no reumáticas (neoplasias hematológicas, enfermedades metabólicas, etc.) pueden afectar al sistema musculoesquelético simulando un proceso o enfermedad reumática. Una buena historia clínica nos ayudará a discernir entre ambas (Tabla II).

“Para diagnosticar una ER se debe pensar en ella”, por lo que es imprescindible contar con determinados conocimientos y habilidades que hagan sospecharla, para mediante una anamnesis y exploración adecuadas, orientar las exploraciones complementarias necesarias y poder realizar un diagnóstico precoz que, sin duda, mejorará el pronóstico, al permitir iniciar, cuanto antes, el tratamiento específico.
En este trabajo abordaremos, inicialmente, la recogida sistemática de síntomas mediante una anamnesis dirigida y, a continuación, la exploración física reumatológica.
Anamnesis
Motivo de consulta
El motivo principal de la consulta debe reflejarse de forma concisa, como por ejemplo: “artralgias de larga evolución”, “inflamación rodilla”, “fiebre y exantema a estudio”, “cojera de reciente comienzo”. De esta forma, puede establecerse, en muchas ocasiones, el síntoma/signo guía de un determinado grupo de patologías musculoesqueléticas.
Antecedentes familiares (AF) (Tabla III)

Aunque en la mayoría de la ER de base inmune, no se ha identificado un gen específico que predisponga a ellas, no es infrecuente que los niños con ER tengan familiares con enfermedades autoinmunes, como: artritis reumatoide, espondiloartritis, tiroiditis, psoriasis, diabetes mellitus, etc. Lo que podríamos denominar como “carga familiar”. En este sentido, debemos recalcar que la presencia de espondiloartritis o psoriasis en un familiar de primer grado, constituye un criterio para la clasificación de las artritis crónicas de niños y adolescentes.
Por otro lado, determinadas enfermedades presentan patrones de herencia más claros, como: los síndromes de fiebre periódica (a la cabeza, la fiebre mediterránea familiar), la osteogénesis imperfecta, el síndrome de Marfan, enfermedad de Ehlers-Danlos, etc. En estos casos, resulta de utilidad realizar un árbol genealógico.
También, se debe tener en cuenta que muchas enfermedades genéticas tienen expresión clínica en el sistema musculoesquelético y, en ocasiones, simulan una enfermedad reumática.
Antecedentes personales (Tabla IV)

• Traumatismos y sobrecargas: siempre debemos tener en cuenta, determinados antecedentes que pueden orientar el diagnóstico, como por ejemplo: el antecedente traumático o de sobrecarga mecánica en pacientes con dolor, tumefacción o limitación de la movilidad de una articulación. Pero, por otro lado, también se debe considerar como posible, que una caída sea consecuencia y no causa de una artropatía ya existente.
En casos de piomiositis, es frecuente el antecedente de sobrecarga física o traumatismos repetidos, días antes de la infección muscular en pacientes jóvenes sin otros antecedentes relevantes.
• Infecciones: el antecedente de determinadas infecciones, como por ejemplo, gastroenteritis entre 1 y 4 semanas antes de la aparición de artritis aguda, puede orientarnos a una artritis reactiva.
La infección estreptocócica por estreptococo beta hemolítico del grupo A, es el agente responsable de la fiebre reumática que hoy difícilmente se observa, pero que no debe olvidarse. En la actualidad, es más frecuente la artritis reactiva posestreptocócica y quizás episodios de uveítis anterior (“uveítis reactiva posestreptocócica”).
El antecedente de un proceso infeccioso respiratorio de vías altas se constata con frecuencia en casos de sinovitis transitoria de cadera (STC).
Antecedentes epidemiológicos
La HCR debe recoger siempre aspectos epidemiológicos que pueden ser de interés para el diagnóstico de determinadas patologías, debiendo interrogar por el contacto con enfermos de tuberculosis, contacto con animales, consumo de lácteos y derivados sin higienizar, picaduras de garrapatas o mosquitos, viajes recientes, etc.
La tuberculosis todavía sigue declarándose en España y puede afectar al sistema musculoesquelético en forma de artritis séptica, osteomielitis y espondilodiscitis. Por este motivo, siempre se debe interrogar por la posibilidad de contacto del paciente con enfermos de TB en su ámbito cercano.
La brucelosis es una antropozoonosis que puede transmitirse por el contacto con animales enfermos o bien por el consumo de productos lácteos sin higienizar (leche cruda no pasteurizada). Esta enfermedad produce fiebre prolongada y compromiso articular con frecuencia, ya sea como artromialgias o bien como artritis, espondilitis, sacroileítis, etc. En España, se siguen declarando casos de brucelosis humana, pero de forma anecdótica en la actualidad.
La enfermedad de Lyme es una borreliosis que es transmitida por picaduras de garrapatas y que cursa con frecuencia con: artritis, exantema característico y compromiso neurológico, entre otros. Es endémica en determinadas latitudes y debe tenerse en cuenta en pacientes procedentes de estas latitudes, sobre todo, si han sufrido picaduras de garrapata.
La fiebre Chikungunya es una enfermedad vírica transmitida al ser humano por mosquitos infectados. Cursa con: fiebre, afectación del estado general, exantema y artromialgias intensas. La enfermedad se da en África, Asia y el subcontinente indio. Recientemente, los vectores de la enfermedad se han propagado a Europa y América, por lo que debe tenerse en cuenta en pacientes con fiebre, exantema y clínica articular.
Otras enfermedades o procesos
El antecedente de dolor abdominal recurrente y rectorragias en un paciente con patología articular, puede ponernos en alerta de la existencia de una enfermedad inflamatoria intestinal (diagnosticada o no) en la que es habitual la presencia de manifestaciones musculoesqueléticas.
Los pacientes con psoriasis cutánea pueden desarrollar artritis crónica durante su evolución en un porcentaje variable de casos, por lo que se debe tener en cuenta en la anamnesis de pacientes en estudio por artropatía. Dado que, en ocasiones, no está diagnosticada, se debe interrogar por la presencia de lesiones características.
Uveítis: el antecedente de inflamación ocular en un paciente con síntomas musculoesqueléticos, puede ponernos sobre la pista de una artropatía inflamatoria crónica, sarcoidosis, enfermedad de Behcet, etc.
Otras enfermedades sistémicas, ya sean autoinmunes, como LES, o autoinflamatoiras, como: la artritis sistémica juvenil, síndromes hereditarios de fiebre periódica (fiebre mediterránea familiar y otras), criopirinopatías (síndrome CINCA), etc., pueden presentar patología musculoesquelética durante su evolución.
En niños con hiperlaxitud, son más frecuentes los cuadros de dolor crónico musculoesquelético.
Historia actual (Tabla V)

La historia del proceso actual contempla, tanto los síntomas musculoesqueléticos relacionados con la inflamación (dolor, tumefacción, limitación de la movilidad y pérdida de función), como las manifestaciones asociadas.
1. Dolor y sus características: se debe interrogar sobre la localización, irradiación, tipo (urente, sordo, quemante, etc.), intensidad, ritmo horario (matutino, nocturno), duración, tiempo de evolución, cadencia, relación con la actividad física, etc. También sobre síntomas acompañantes, como: aumento de temperatura local, fiebre enrojecimiento, etc.
Por lo general, las enfermedades inflamatorias articulares (AIJ) no se caracterizan por el dolor como síntoma principal, siendo más frecuente que se refiera por parte de los padres: hinchazón y/o limitación de movilidad de un miembro, o bien, que el niño rehúse o rechace la realización de actividades habituales.
Cuando el dolor es intenso o desproporcionado a la tumefacción articular, debemos plantear la posibilidad de una artritis séptica o un proceso tumoral; en estos casos, el ritmo del dolor y la presencia de síntomas acompañantes, como fiebre y afectación del estado general, nos pondrán sobre la pista.
El dolor musculoesquelético (Fig. 1 y Tabla VI) generalizado sin signos de alarma es característico de los síndromes de sensibilización central (p. ej., fibromialgia juvenil) o de amplificación del dolor.

Figura 1. Esquema del abordaje del dolor musculoesquelético en reumatología pediátrica.

Dolor localizado de ritmo mecánico o no definido, puede corresponder tanto a sobrecarga mecánica por sobreuso, como a dolores del crecimiento, osteocondrosis (enfermedad de Sever, Osgood Schlatter, etc.) por lo general, procesos benignos.
2. Tumefacción o hinchazón: la tumefacción articular no siempre es muy evidente o afecta a articulaciones pequeñas o poco habituales y puede pasar desapercibida, por lo que la exploración física será de mayor utilidad para ponerla de manifiesto. Hay que tener en cuenta que la tumefacción puede afectar a tejidos periarticulares, vainas tendinosas y ser interpretado erróneamente como una artritis.
No debemos olvidar que las tenosinovitis se objetivan con frecuencia en la AIJ asociada a la artritis de carpos, tobillos y pies. Cuando se refiere al retropié, puede tratarse de una tendinitis aquílea, característica de la artritis idiopática juvenil forma “artritis y entesitis”.
La tumefacción que se extiende más allá de una articulación puede corresponder a una dactilitis (“dedo en salchicha”) y es característica de determinadas formas de AIJ.
En ocasiones, puede objetivarse edema periarticular, acompañando a un proceso inflamatorio articular que puede corresponder a un linfedema.
3. La limitación de la movilidad de una articulación o un miembro y la incapacidad para realizar actividades habituales. Pueden deberse, tanto a un proceso banal o inespecífico, como a un proceso infeccioso (miositis, artritis séptica, osteomielitis), como a un inflamatorio crónico (AIJ) o incluso tumoral (leucemia linfoblástica aguda, sarcoma de Ewing, etc.).
La limitación de la movilidad puede deberse también a debilidad, que cuando afecta de forma proximal a miembros es característica de las miopatías inflamatorias (DMJ), la presencia de síntomas y signos característicos nos orientarán al diagnóstico.
La cojera es frecuente en la edad pediátrica y representa un mecanismo de protección contra el dolor. En ocasiones, es transitoria y precedida, por lo general, de una infección de vías respiratorias altas (sinovitis transitoria de cadera). Cuando la sinovitis persiste más de 1-2 semanas o se acompaña de episodios de cojera previa, estamos obligados a descartar una enfermedad de Perthes.
La presencia de fiebre asociada a la cojera nos obligará a descartar una artritis séptica/osteomielitis.
No debemos olvidar que la cojera puede ser expresión de una coxitis inflamatoria en un paciente con artritis idiopática juvenil. Aunque la enfermedad puede debutar afectando a la cadera, esto es poco frecuente y, por lo general, la coxitis se presenta en el curso evolutivo de la misma, habitualmente en varones mayores de 9-10 años con la forma “artritis y entesitis”.
Es importante tener en cuenta que no siempre la cojera o limitación de la marcha es debida a un problema de cadera, como ocurre con frecuencia en la inflamación de articulaciones de miembros inferiores de la artritis idiopática juvenil. En este sentido, cuando un niño cojea o rehúsa caminar o mantener la postura, se debe contemplar también la posibilidad de un problema de columna, como por ejemplo, una discitis que, por lo general, afecta a niños de 2-5 años. La exploración física nos ayudará a valorar estas opciones una vez sospechadas.
Las contracturas en flexión (“flexos”), por lo general no dolorosas, son relativamente frecuentes en la artritis idiopática juvenil, cuando se afecta el codo o la rodilla, ya sea en su inicio como durante su evolución. En ocasiones, la enfermedad se descubre estudiando un flexo y pueden pasar inadvertidos si son leves.
Una forma rápida de valorar la limitación de forma global, es empezar preguntando: si el niño presenta dificultad para vestirse sin ayuda, o subir o bajar escaleras (si antes lo hacía).
4. Incapacidad funcional: la patología reumática se acompaña, por lo general, de limitación funcional y su valoración ha de tener en cuenta el grupo etario del paciente: en el lactante, se observa una disminución de la movilidad espontánea, llanto desencadenado por determinados movimientos o una postura antiálgica durante el sueño; en los preescolares, la limitación puede hacerse patente por la alteración de la actividad normal al vestirse, al subir o bajar escaleras, al realizar actividades deportivas, etc.
5. Rigidez: la presencia de rigidez o entumecimiento matutino o tras periodos de reposo o inactividad más o menos prolongados, es característica de la patología inflamatoria articular (artritis idiopática juvenil), pudiendo llegar a ser el signo más importante de las ER. Cuando se asocia a dolor o tumefacción, decimos que el dolor es de ritmo inflamatorio en contraposición al de ritmo mecánico, que no se acompaña de rigidez y se presenta después de la realización de actividad física o al final del día. Por este motivo, es muy importante preguntar siempre si el niño tiene: rigidez, entumecimiento o rehúsa moverse al levantarse por la mañana o tras periodos de reposo. Los niños mayores suelen referirla, como: molestias vagas, sensación de cansancio, debilidad muscular, torpeza, agarrotamiento, etc.
6. Síntomas generales: la presencia de síntomas, como: fiebre, malestar general, decaimiento, debilidad, pérdida de peso, etc., pueden presentarse asociados a patología reumática, como: AIJ sistémica, LES, vasculitis o enfermedad de Kawasaki, pero estamos obligados a sospechar una infección o un proceso tumoral.
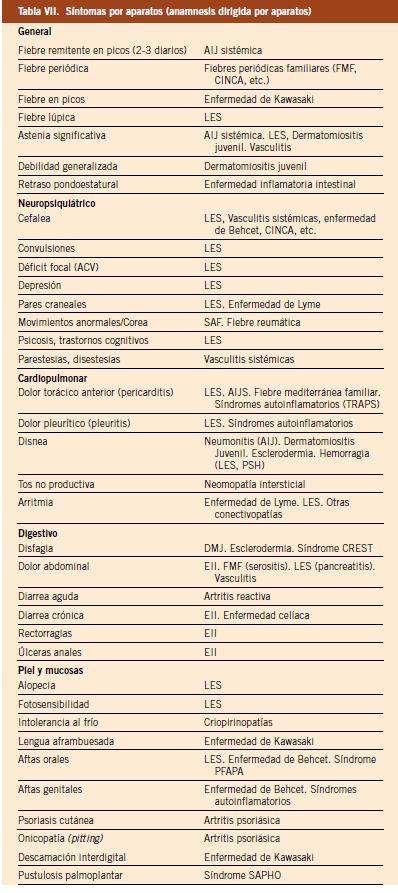
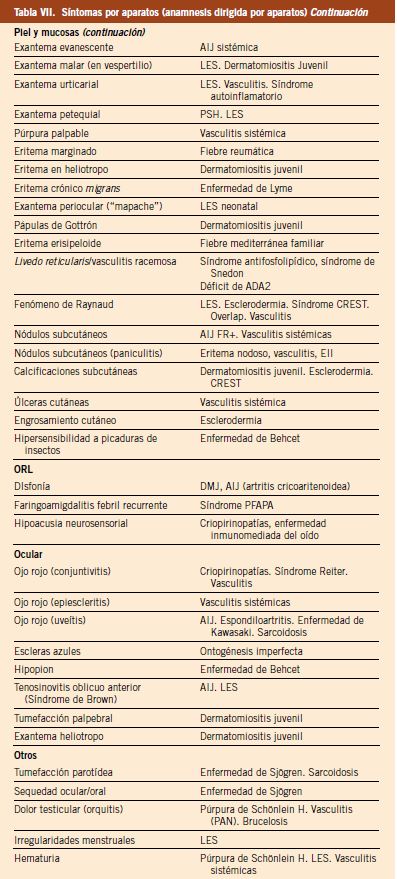
7. Síntomas por aparatos (anamnesis dirigida por aparatos): la anamnesis en Reumatología debe ir dirigida a la búsqueda de síntomas que, si están presentes, puedan orientarnos al diagnóstico de un determinado proceso reumatológico, ya sea este de naturaleza inflamatoria o no; así, vemos como, en la patología de base inmune, pueden estar presentes de forma aislada o combinada: fiebre, astenia, artralgias, mialgias, debilidad, rigidez matutina, aftas orales, fotosensibilidad, fenómeno de Raynaud, fiebre, etc., que, en muchas ocasiones, el paciente no refiere y que hemos de preguntar de forma más o menos sistemática y dirigida para hacerlas patentes. En la tabla VII, se detallan los principales hallazgos que debemos tener en cuenta para preguntar sobre ellos y confirmar posteriormente en la exploración, si procediera.
Exploración física en reumatología pediátrica
Una vez realizada la anamnesis, continuaremos con el examen musculoesquelético, realizado de forma sistemática a través de la inspección, palpación y examen de la movilidad articular, para lo que tendremos en cuenta los siguientes apartados:
• Estática y actitud articular: con el paciente desnudo, se valorarán: la simetría en los relieves óseos, la morfología de las articulaciones, las masas musculares, la actitud postural, las posibles desviaciones del raquis y/o de las extremidades o la dismetría de las extremidades inferiores. La presencia de contracturas en flexión (“flexos”) suele ponernos en alerta de patología articular inflamatoria o infecciosa, especialmente en: codo, muñeca, cadera y rodilla…
• Signos inflamatorios: los signos clásicos de la inflamación deben explorarse en busca de tumefacción articular o periarticular, rubor y calor. A continuación, valoraremos la presencia de derrame articular en articulaciones accesibles.
• Deformidades: una articulación deformada indica, por lo general, un proceso de larga duración. Pueden ser congénitas o adquiridas.
• Puntos dolorosos: la presencia de dolor en la exploración musculoesquelética, habitualmente tiene carácter patológico y debe valorarse siempre. Se valorará, especialmente: dolor articular, periarticular, músculos, fascias, tendones y sus inserciones. Hay que tener en cuenta que en muchos niños con enfermedades reumáticas, no se refiere ni se objetiva dolor en la exploración, siendo más habitual que el niño rehúse realizar determinados movimientos o constatar retirada al palpar o mover una articulación o estructura relacionada.
En ocasiones, se objetiva en la exploración muchas áreas dolorosas que puede traducir: tensión muscular, procesos de amplificación del dolor o un síndrome de sensibilización central (fibromialgia y patologías relacionadas). Un dolor a la exploración desproporcionado o acompañado de signos de compromiso sistémico, nos hará sospechar un proceso infeccioso osteoarticular o neoplásico con expresión musculoesquelética.
• Movilidad articular: se evaluará la movilidad, tanto activa como pasiva, en busca de cualquier tipo de limitación, que puede ser parcial o total (anquilosis) y acompañarse o no de dolor. Si por el contrario se objetiva movilidad excesiva, podremos hablar de hiperlaxitud articular generalizada, cuando se cumplen determinados criterios (Tabla VIII).

No hay que olvidar que la presencia de hiperlaxitud generalizada puede ser una manifestación de síndromes, como: Marfan, Ehlers-Danlos, etc., que asocian un fenotipo particular.
• Limitación funcional: la alteración de la capacidad funcional es consecuencia directa del dolor o tumefacción que provoca el proceso articular. Cuando se afectan las extremidades inferiores, en la mayoría de los casos, el niño presenta cojera o imposibilidad para apoyar la extremidad, dejando de realizar actividades habituales, como: jugar, correr, montar en bicicleta o cualquier actividad deportiva. Cuando se afectan las extremidades superiores, es más difícil de apreciar, debiendo valorarse las actividades manuales relacionadas con el aseo personal o el vestirse. En el caso de los lactantes, se puede observar disminución de la movilidad en la cuna o posturas inusuales durante el sueño.
La exploración física debe ser sistemática y, para ello, nos resultará útil establecer un orden como el propuesto.
Columna cervical
En pacientes con AIJ, sobre todo en las formas con afectación poliarticular, no es infrecuente que se afecte la columna cervical; para valorar esta posibilidad, empezaremos examinando la flexión, extensión, rotaciones y lateralización del cuello con el paciente de pie o sentado, pidiendo al niño que mire hacia el techo, luego a su ombligo y, finalmente, hacia los lados y hacia atrás.
Un signo frecuente de afectación cervical es la limitación, por lo general no dolorosa, de la extensión para sus últimos grados (Fig. 2).

Figura 2. Limitación de la extensión cervical en paciente con AIJ poliarticular evolucionada.
Así mismo, es posible observar como el niño gira el tronco cuando se le pide que mire hacia atrás en vez de mover el cuello solamente.
A continuación, valoraremos mediante palpación las masas musculares del cuello en búsqueda de contracturas dolorosas o no. En este momento, podemos aprovechar para buscar adenopatías.
Finalmente en decúbito supino, se explora la rotación cervical, moviendo la cabeza 90º tanto hacia la derecha como hacia la izquierda (Fig. 3).

Figura 3. Exploración de las rotaciones cervicales con el paciente en decúbito supino.
Articulación temporomandibular (ATM)
La ATM se afecta con cierta frecuencia en niños con artritis idiopática juvenil, pero puede pasar desapercibida, sobre todo, inicialmente.
Los principales signos de afectación ATM se exponen en la tabla IX y en las figuras 4-6.


Figura 4. Asimetría de la apertura oral en paciente con AIJ oligoarticular.

Figura 5. Exploración de la apertura oral. Se considera normal la introducción de, al menos, tres dedos.
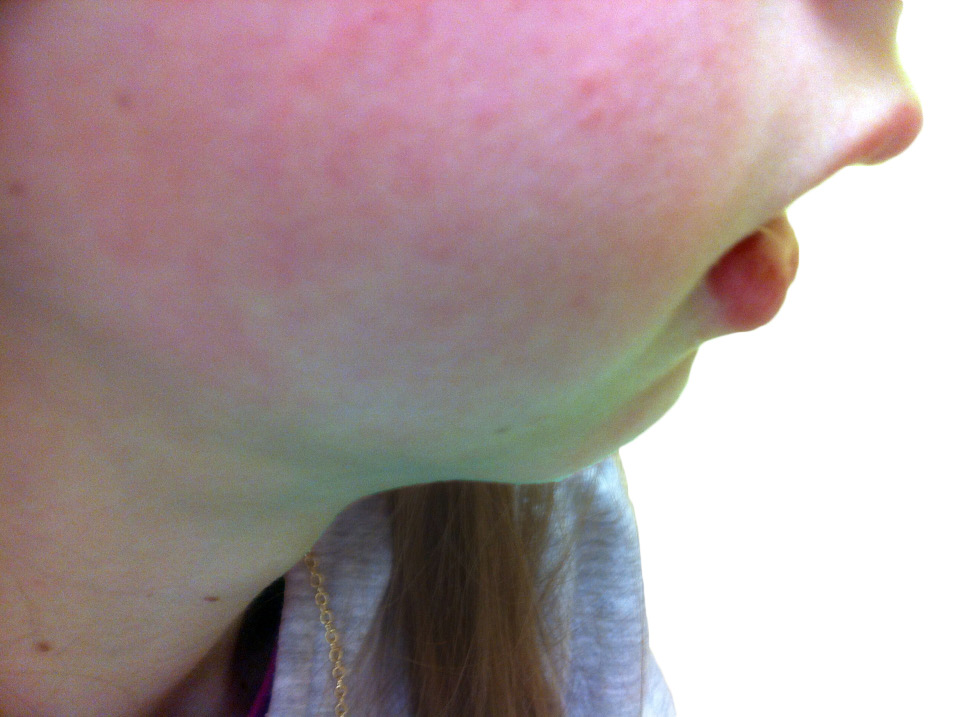
Figura 6. Retrognatia en paciente con AIJ poliarticular evolucionada.
Articulación esternoclavicular
En ocasiones, puede afectarse la articulación esternoclavicular (Fig 7) en procesos inflamatorios o infecciosos óseos o articulares. La artritis suele pasar desapercibida, por lo que debe examinarse dentro de la sistemática que se establezca.
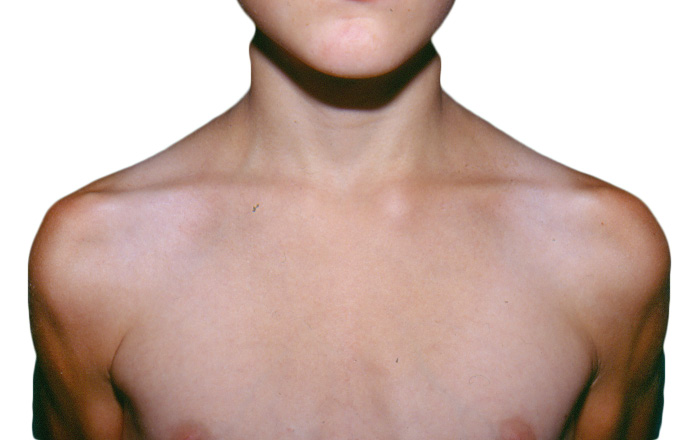
Figura 7. Tumefacción articulación esternoclavicular izquierda en paciente con Artritis y entesitis.
Hombro
La articulación del hombro se afecta con poca frecuencia en las artropatías crónicas, pero debe explorarse sistemáticamente. La inflamación de la articulación glenohumeral puede ponerse de manifiesto por la presencia de tumefacción en la cara anterior del hombro que puede resultar dolorosa a la palpación. No debemos olvidar que este hallazgo puede corresponder también a la presencia de tenosinovitis o bursitis, que es poco frecuente en pacientes con artritis crónicas, pero que hay que tener en cuenta.
La movilidad debe evaluarse pidiendo al niño que eleve los brazos hacia delante, hacia atrás y hacia los lados (abducción). Acto seguido, se le invita a que toque el hombro opuesto de cada brazo, tanto por delante como por la espalda (rotaciones externa e interna).
También puede explorarse la movilidad pasiva del hombro mediante la flexión del antebrazo sobre el brazo 90º, dirigiendo el antebrazo y la mano hacia fuera y hacia dentro (“tocar el ombligo”) y, a continuación, levantando el codo (brazo en abducción de 90º) llevar la mano tanto hacia arriba como hacia abajo (rotación externa e interna).
El signo más frecuente de compromiso articular es la limitación, en ocasiones dolorosa, de la rotación interna, tanto activa como pasiva (Fig. 8).

Figura 8. Exploración rotación interna del hombro.
Codo
La articulación del codo se afecta con frecuencia, pero puede pasar desapercibido si la artritis es leve y no se explora adecuadamente.
La sinovitis puede sospecharse por la presencia de tumefacción (Fig. 9) en la inspección, o bien, palpando la articulación radio-humeral en la parte posterior del codo (surco retroolecraniano externo), mientras se realiza un movimiento de pronosupinación de la mano.
Se han de explorar, tanto la flexoextensión como pronosupinación para valorar si existe algún grado de dolor y limitación de la movilidad.
Con frecuencia, el primer signo de artritis del codo es la limitación de la extensión completa para sus últimos grados (flexo), que puede ser dolorosa o no. Los niños pequeños suelen tener tendencia a la hipermovilidad de los codos y, por tanto, un codo aparentemente normal, pero que no hiperextiende unos grados, puede ser el primer signo de una sinovitis, sobre todo, si el hallazgo es asimétrico.
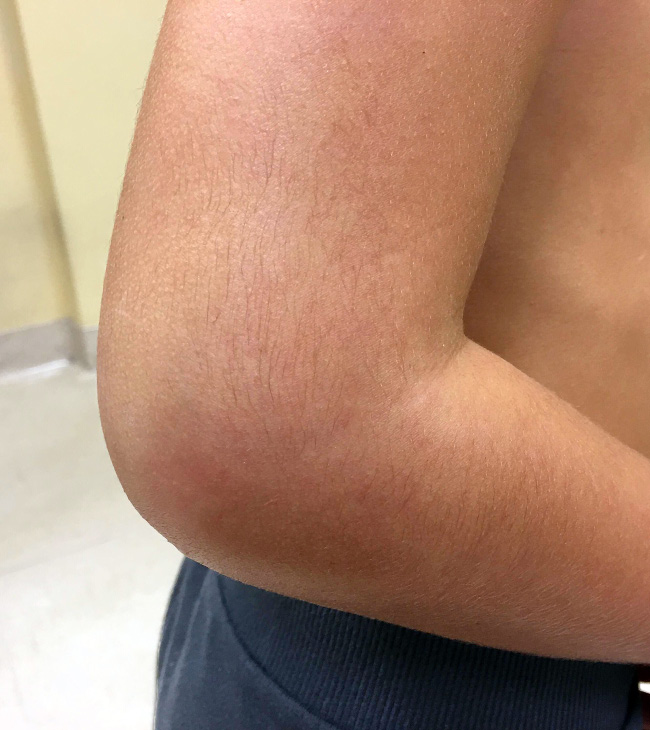
Figura 9. Tumefacción marcada codo derecho y partes blandas adyacentes en paciente con artritis séptica.
Muñeca
La sinovitis de la articulación radiocarpiana y/o vainas tendinosas adyacentes es muy frecuente en pacientes con AIJ y se puede detectar mediante la simple inspección en forma de abultamientos o tumoraciones de consistencia blanda, por lo general, en su cara dorsal (Fig. 10).
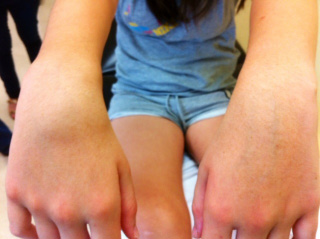
Figura 10. Tumefacción por tenosinovitis en carpo en paciente con AIJ poliarticular.
No obstante, el compromiso inflamatorio suele hacerse patente de forma más precoz al palpar todo el carpo rodeando con el pulgar y tercer dedo del explorador ambas muñecas y observar cómo se produce dolor o una maniobra de retirada al presionar ligeramente (Fig. 11).
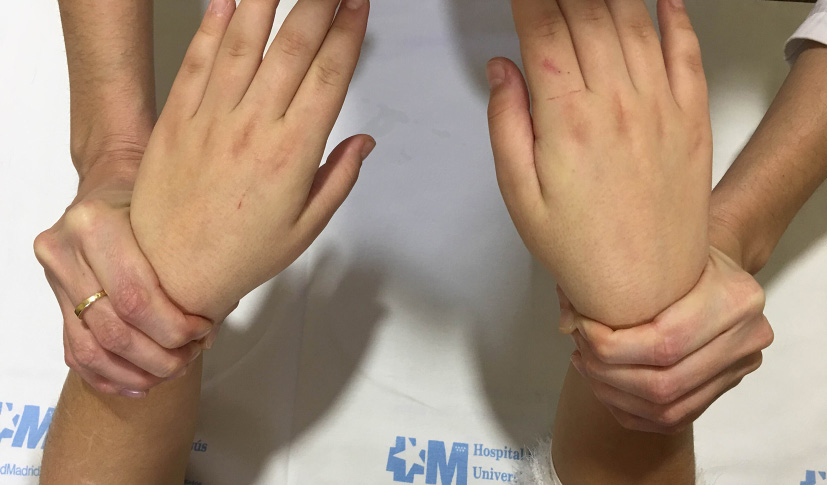
Figura 11. Compresión simultánea de las muñecas para explorar sinovitis en carpos.
A continuación, se evaluará la flexoextensión. Para ello, es útil pedir que el niño con los codos flexionados y apoyados sobre una mesa, intente mantener juntas las palmas de las manos en actitud de oración (Fig. 12), y descender el borde de las manos para tocar la mesa. Esta maniobra puede poner de manifiesto, de forma precoz, la inflamación de un carpo que de otra manera podría pasar desapercibido.

Figura 12. Actitud en “rezo” para evaluar extensión de carpos y articulaciones de los dedos.
Posteriormente, se juntarán los dorsos de las manos dirigiendo los dedos hacia abajo en vertical. La movilidad pasiva debe ser de 90º tanto para la flexión como para la extensión. También, deben explorarse los movimientos de lateralización cubital y radial, que en circunstancias normales son de 35 y 45º, respectivamente.
Mano
Las articulaciones de la mano se afectan con frecuencia en las artropatías crónicas. Mediante la simple inspección, podemos poner de manifiesto la presencia de tumefacción articular en los dedos. La presencia de dactilitis (Fig. 13) es muy sugerente de una enfermedad inflamatoria crónica; en ocasiones, pasa desapercibida si no es muy manifiesta, dado que, por lo general, no resulta dolorosa.

Figura 13. Dactilitis 4 dedo mano derecha en paciente con AIJ psoriásica.
También, mediante la inspección pueden ponerse de manifiesto la presencia de contracturas articulares y deformidades en metacarpofalángicas o interfalángicas que, por lo general, traducen procesos evolucionados (Fig. 14).

Figura 14. Contracturas en flexión de interfalángicas proximales en paciente con AIJ poliarituclar.
También, se deben inspeccionar las uñas en busca de onicolisis o piqueteado que pueden acompañar a la forma de artropatía psoriásica (Fig. 15).

Figura 15. Pitting ungueal en paciente con AIJ psoriásica.
Es recomendable pedir al niño que haga el puño (Fig. 16), para valorar si existe tumefacción o limitación de la movilidad de las metacarpofalángicas y acto seguido se le invita a que nos enseñe las uñas, flexionando las interfalángicas proximales y distales totalmente, mientras permanecen extendidas las metacarpofalángicas (maniobra de Bunnell).
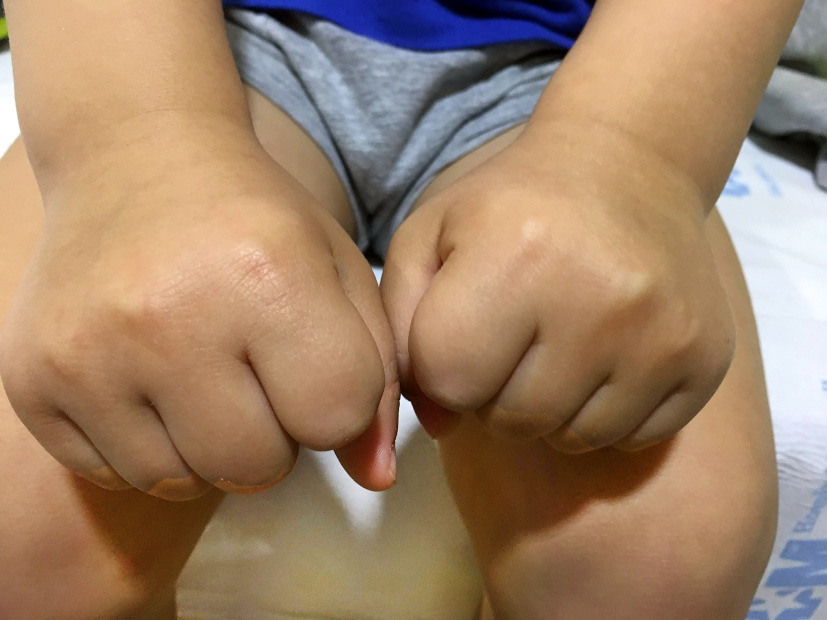
Figura 16. Maniobra del puño para evaluar sinovitis de metacarpofalángicas e interfalángicas.
Las articulaciones metacarpofalángicas se evalúan inicialmente de forma conjunta, ejerciendo una compresión lateral entre la 2ª y la 5ª. Su movilidad en flexión es de unos 80º y en extensión de 45º.
Las articulaciones interfalángicas se exploran presionando con el dedo pulgar e índice sobre las caras laterales de la articulación. La flexión normal es de 90º.
La hipermovilidad del primer y del quinto dedo por laxitud es uno de los criterios diagnósticos de hiperlaxitud.
Cadera
El compromiso de la articulación coxomeforal puede abarcar, desde procesos transitorios, como una sinovitis transitoria de cadera, una enfermedad de Perthes o, incluso, un proceso séptico o tumoral, pero no debemos olvidar que esta articulación puede afectarse en niños con AIJ, por lo general, niños mayores en la forma artritis y entesitis (lo que podríamos considerar una espondiloartritis juvenil). Lo más habitual, es que se afecte durante el curso evolutivo de la misma, siendo poco frecuente que la enfermedad debute con una coxitis.
La limitación dolorosa de la rotación interna de la cadera suele ser el signo más habitual en caso de sinovitis. Sin embargo, una actitud en flexión y rotación externa, puede objetivarse como un signo de alarma precoz en la artritis séptica de cadera (Fig. 17).
La flexión debe explorarse intentando llevar la rodilla hasta el pecho (rango normal 120-135º). Las rotaciones se exploran con la cadera, rodilla y tobillo flexionados 90º, aproximando o separando la pierna de la línea media.

Figura 17. Cadera: maniobra de rotación externa (pie hacia dentro) e interna (pie hacia fuera).
Conviene realizar la maniobra de rolling (Fig. 18), que evalúa la rotación sobre su eje del miembro inferior totalmente extendido; para ello, se sitúan las manos por encima y por debajo de la rodilla, haciendo rotar la extremidad hacia dentro y hacia fuera. En la artritis séptica de cadera, se objetivará una limitación dolorosa de las rotaciones a diferencia de la sinovitis transitoria de cadera que no suele doler.

Figura 18. Maniobra de “rolling” para valorar rotación de cadera, que característicamente no resulta dolorosa en sinovitis transitoria de cadera a partir de los 30º, a diferencia de otros procesos.
Acto seguido, en decúbito supino, se explorarán tanto la extensión como las rotaciones, para ello, se fijará la pelvis con una mano y con la otra se elevará la pierna en extensión completa (el rango normal es de 30º). A continuación, se evalúan las rotaciones flexionando las piernas 90º y aproximando y separando cada pierna con respecto a la línea media (Fig. 19).

Figura 19. Maniobra de rotación interna de ambas caderas.
Articulación sacroilíaca y columna lumbar
Las articulaciones sacroilíacas suelen ser las grandes desconocidas en la patología musculoesquelética, pero hay que poner de manifiesto que, también, pueden presentar inflamación en determinadas formas de AIJ, manifestado, por lo general, como dolor lumbosacro de ritmo inflamatorio y/o limitación de la movilidad lumbar.
En primer lugar, se debe explorar palpando la región lumbosacra en busca de puntos dolorosos sobre el área sacroilíaca. Con frecuencia, puede provocarse dolor en este área en niños y, sobre todo, adolescentes con procesos miofasciales o de amplificación del dolor, pero también puede tratarse de una sacroileítis subclínica en un niño con una artropatía inflamatoria crónica.
Las maniobras clásicas para explorar la sinovitis incluye: la maniobra de FABERE (Flexión, Abducción, Rotación Externa), en la que estando el niño en decúbito supino, se flexiona la rodilla para apoyar el tobillo en la rodilla del miembro opuesto. Apoyaremos una mano sobre la espina ilíaca ipsilateral, al mismo tiempo que realizamos una suave flexión sobre la rodilla del lado examinado. La prueba es positiva, cuando genera dolor en la región sacroilíaca homolateral (Fig. 20).

Figura 20. Prueba de FABERE. Flexión abducción y rotación externa, fijando la pelvis para evaluar sacroileítis.
También es clásica, aunque menos usada en niños, la maniobra de Menell: en decúbito lateral, con la cadera y la rodilla del lado supuestamente afectado en hiperflexión, y la pierna del lado opuesto, proyectada en hiperextensión hacia atrás con la rodilla flexionada. Es positiva, cuando se genera dolor al comprimir la línea interarticular sacroilíaca al tiempo que se realiza una hiperextensión del muslo.
La movilidad lumbar se explora pidiendo al paciente que intente flexionar el tronco para intentar llevar las palmas totalmente extendidas al suelo, si lo consigue, sin duda, estaremos ante un caso de hiperlaxitud y habrá que comprobar si también otras articulaciones son laxas para diagnosticar un síndrome de hiperlaxitud.
Si, por el contrario, se objetiva una limitación “significativa” en la flexión, hay que valorar la posibilidad de un proceso inflamatorio crónico que esté afectando o ha afectado al esqueleto axial (artritis y entesitis o espondiloartropatías juveniles), para lo cual se realizará el test de Schöber que, además, es útil para monitorizar evolutivamente al paciente.
En niños mayores de 6 años, valorar la limitación de movimientos en la región lumbosacra mediante el Test de Schöber modificado. Con el paciente en bipedestación, se traza una línea a la altura de las espinas ilíacas; a continuación, se marcan dos líneas paralelas a esta, una 5 cm por debajo y otra 10 cm por encima (Fig. 21).

Figura 21. Test de Schöber modificado. Medición previa a la flexión máxima del raquis hacia delante.
El niño realizará una flexión del tronco para intentar alcanzar el suelo con los dedos de las manos. En este momento, se mide la diferencia de distancia entre el punto superior e inferior entre la posición erecta (15 cm) y en flexión máxima (21-22 cm). En niños mayores de 6 años, se considera patológica una diferencia inferior a 6 cm. Además, debemos explorar la flexión lateral del raquis, evaluando la distancia desde el tercer dedo de la mano hasta el suelo, de forma periódica.
Rodilla
El examen de la rodilla comienza con la inspección del paciente en decúbito supino para poner de manifiesto: tumefacción articular o periarticular, atrofia de masas musculares (cuádriceps) o una actitud en flexión (“flexo”) que, casi siempre, serán patológicas (Fig. 22).

Figura 22. Actitud en flexo de rodilla izquierda en paciente con AIJ oligoarticular.
Se valorará la existencia de dolor a la palpación o aumento de temperatura, como signos inflamatorios habitualmente presentes en caso de sinovitis aguda.
La observación de un abultamiento, a veces, doloroso en el área que corresponde a la inserción del tendón rotuliano en la tuberosidad anterior de la tibia, corresponde, por lo general, a una osteocondritis de Osgood Schlatter.
La presencia de dolor en el polo inferior de la rótula en el área de la inserción proximal del tendón rotuliano, puede deberse a una osteocondritis de Sinding Larsen Johanson.
No debemos olvidar que las inserciones del tendón rotuliano pueden afectarse en caso de entesitis en el contexto de una enfermedad inflamatoria, como la artritis entesitis o una espondiloartritis juvenil.
El dolor en el tendón rotuliano que se incrementa significativamente al realizar una extensión resistida de la rodilla, nos ha de hacer pensar en una tendinitis rotuliana por sobreuso en pacientes deportistas.
A continuación, se comprimen con la mano los fondos de saco laterales de la rodilla para desplazar el líquido sinovial existente hacia el aspecto contralateral, comprobando si se produce abombamiento, en cuyo caso hablaremos de un signo de la oleada positivo, que traduce la presencia de exceso de líquido intraarticular, habitual ante una sinovitis.
Con el mismo objetivo que en la maniobra anterior, se recomienda presionar sobre la rótula con el dedo pulgar, mientras se exprime el fondo de saco subcuadricipital con los dos primeros dedos de la otra mano, de esta manera, se desplazará el líquido sinovial presente hacia la región infrarotuliana. Si el observador siente la fluctuación de la rótula bajo el dedo del explorador, el “signo del peloteo rotuliano o del balón” es positivo y hay derrame sinovial.
Tobillo y pie
El tobillo y pie se exploran buscando tumefacción difusa (Fig. 23) o localizada en las diferentes articulaciones: tibioastragalina, subastragalina, mediotarsiana, metatarsofalángicas e interfalángicas, y en las vainas de los tendones, tanto de la cara anterior (principalmente, tibial anterior) como medial (principalmente, tibial posterior) y lateral (principalmente, peroneos), que nos orientará a la posibilidad de un proceso inflamatorio o infeccioso.
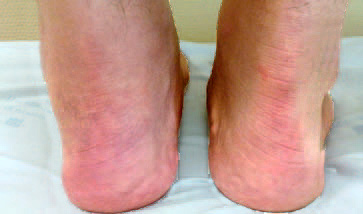
Figura 23. Tumefacción difusa retropié, tendinitis aquílea. Paciente con artritis entesitis HLA B27+.
También, a simple vista, puede ponerse de manifiesto una dactilitis que adopta el aspecto de “dedo en salchicha” y que corresponde a un proceso inflamatorio de una articulación del dedo que se extiende más allá de la misma, y que suele observarse en niños con AIJ (mayoritariamente en la forma artritis y psoriasis) (Fig. 24).

Figura 24. Dactilitis (“dedo en salchicha”) en paciente con AIJ psoriásica.
La movilidad activa y pasiva del tobillo se afecta en caso de sinovitis o tenosinovitis. Debemos valorarla adecuadamente teniendo en cuenta:
• Articulación tibioastragalina: la flexión dorsal habitual es de 20º y la plantar 45º.
• Articulación subastragalina: lo normal son 30º tanto para la inversión como para la eversión del pie.
• El tarso se valora mediante la rotación del antepié sobre el retropié.
• Las articulaciones pequeñas del antepié se exploran conjuntamente comprimiendo lateralmente las articulaciones metatarsofalángicas y las interfalángicas.
• Acto seguido, observaremos al niño caminando de puntillas y talones durante unos metros.
• Para finalizar buscamos puntos dolorosos en el tendón de Aquiles y su inserción en el calcáneo, y en las inserciones de la fascia plantar (tuberosidad del calcáneo, cabezas de metatarsianos y base del quinto metatarsiano), que resultan dolorosos en caso de entesitis, característica de la forma artritis y entesitis de la AIJ y espondiloartritis juveniles (Fig. 25).
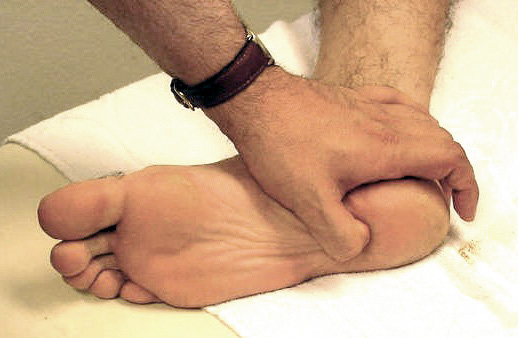
Figura 25. Exploración de la entesitis calcánea de la fascia plantar. Dolorosa en fascitis plantar en pacientes con artritis entesitis.
Otros aspectos en la exploración musculoesquelética
Para finalizar, la exploración física debe contemplar también los siguientes apartados:
• Marcha: siempre debe explorarse la deambulación en búsqueda de cojera por afectación de la cadera u otras articulaciones del miembro inferior. Se debe tener en cuenta que con frecuencia la única manifestación es la claudicación de la marcha.
Determinados patrones de la marcha pueden orientarnos a procesos concretos:
- Inclinación del tronco hacia delante (afectación de la columna dorsolumbar).
- Marcha salutatoria (afectación de la cadera).
- Dificultad marcha en puntillas/talones (afectación de la rodilla y tobillos).
• Alineación del raquis: los trastornos de alineación del raquis deben valorarse en busca de cifosis, escoliosis o hiperlordosis que, en ocasiones, se objetivan en pacientes reumáticos, aunque, por lo general, como patología asociada y no como una manifestación reumática per se.
• Alineación de miembros: aunque ya hemos comentado que la actitud en flexión es frecuente en la patología articular reumática (“flexos”), podemos poner de manifiesto alteraciones asociadas, como: la hiperextensión de codos por laxitud, el cúbito valgo en miembros superiores y el genu valgo, varo o recurvatum en miembros inferiores, por poner algunos ejemplos.
• Longitud de miembros inferiores: mediremos la distancia existente entre la espina ilíaca anterosuperior y el maléolo interno, en búsqueda de dismetrías que son frecuentes en patología articular crónica de miembros inferiores y que, por tanto, debemos monitorizar.
Para facilitar la sistematización exploratoria, se puede utilizar el examen de cribado musculoesquelético para niños pGALS (pediatrics, Gait, Arms, Legs, Spine) (Tabla X), que combina aspectos de inspección, palpación y movilidad en articulaciones periféricas y el raquis.

Resultará también de utilidad, reflejar en un gráfico el compromiso articular, señalando las articulaciones con dolor, con tumefacción y con limitación de la movilidad (Fig. 26).

Figura 26.
Exploración extraarticular en Reumatología
Las ER son mayoritariamente procesos sistémicos y es frecuente el compromiso extraarticular; por lo que la exploración general aporta con frecuencia hallazgos relevantes para su diagnóstico y caracterización. El examen debe hacerse adaptado a cada periodo de la infancia y adolescencia. Además de valorarse el estado general, de las constantes vitales y parámetros de crecimiento y desarrollo, se han de tener en cuenta determinados signos frecuentes o característicos en las ER. A continuación, se exponen los más relevantes.
Fiebre
Ante un niño con fiebre, la primera posibilidad que hay que tener en cuenta es que se trate de un proceso infeccioso, también puede tener un origen neoplásico, pero dependiendo de la presencia de determinados síntomas o signos característicos, puede ser manifestación de una enfermedad reumática.
En la AIJ sistémica, la fiebre cursa en forma de picos que se presentan varias veces al día. Se suele acompañar de exantema evanescente característico y artralgias o artritis. También, son frecuentes las adenopatías, hepatoesplenomegalia y cuadros de serositis.
Ante un paciente con clínica articular y entesopatía (fascitis plantar, tendinitis aquílea, etc.), la presencia de fiebre y diarrea nos hará pensar en la posibilidad de una enfermedad inflamatoria intestinal.
En las conectivopatías, como el LES y la dermatomiositis, y en las vasculitis sistémicas, como la panarteritis nodosa, enfermedad de Kawasaki y enfermedad de Takayasu, es frecuente la presencia de fiebre asociada a sintomatología constitucional y síntomas y signos característicos.
Por último, cuando la fiebre es prolongada o cursa de forma episódica, se debe pensar también en la posibilidad de una enfermedad autoinflamatoria.
Fenotipos peculiares
• Síndrome de Marfan: talla alta, braza excesiva, hiperlaxitud, luxación cristalina, valvulopatía.
• Síndrome de Ehlers-Danlos: hiperlaxitud generalizada, cicatrices características.
• Síndrome de Stickler: miopía magna, patología degenerativa articular, hipoplasia maxilar, fisura palatina, úvula bipartita.
• Criopirinopatías (Síndrome CINCA): abombamiento frontal, prominencias óseas.
Cráneo facial
• Retrognatia: AIJ.
• Asimetría apertura oral: AIJ.
• Tumefacción parotídea: síndrome de Sjögren. Sarcoidosis.
• Abombamiento frontal: síndrome CINCA.
• Hipoplasia maxilar superior: síndrome de Stickler.
• Boca fruncida: esclerodermia.
Piel y mucosas
• Psoriasis: lesiones eritematodescamativas en superficies extensoras. Artritis psoriásica.
• Eritema anular: exantema fotosensible en lupus cutáneo subagudo.
• Pustulosis palmo plantar: síndrome SAPHO.
• Aftas orales: LES (no dolorosas). Enfermedad de Behcet (dolorosas). Síndrome PFAPA (dolorosas). Síndromes autoinflamatorios por déficit de mevalonato cinasa).
• Aftas nasales: LES.
• Aftas genitales: enfermedad de Behcet. Síndromes autoinflamatorios por déficit de mevalonato cinasa.
• Alopecia: LES.
• Labios fisurados: enfermedad de Kawasaki.
• Lengua aframbuesada: enfermedad de Kawasaki.
• Descamación periungueal: enfermedad de Kawasaki.
• Exantemas:
- Exantema evanescente: exantema maculopapuloso asalmonado de predomino en raíz de miembros: AIJ sistémica (Fig. 27).
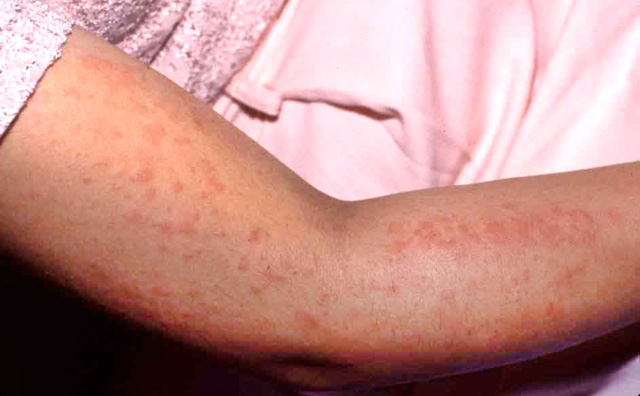
Figura 27. Exantema maculopapular asalmonado evanescente en paciente con artritis idiopática juvenil sistémica.
- Exantema malar en alas de mariposa: LES, dermatomiositis juvenil.
- Eritema en heliotropo: dermatomiositis juvenil (Fig. 28).
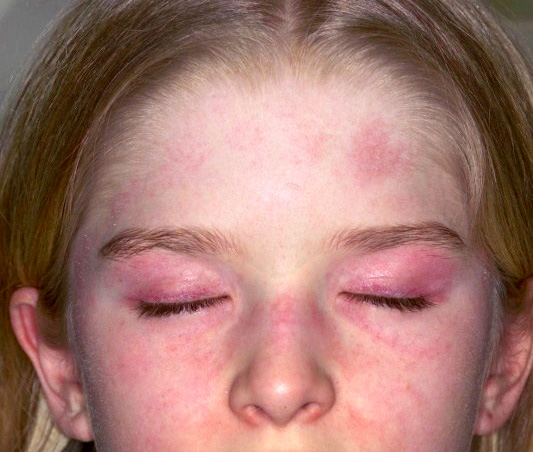
Figura 28. Exantema eritematovioláceo en párpados (eritema en heliotropo) y exantema facial en alas de mariposa. Paciente con dermatomiositis juvenil.
- Púrpurico: púrpura de Schönlein Henoch (Fig. 29). Vasculitis ANCA +.
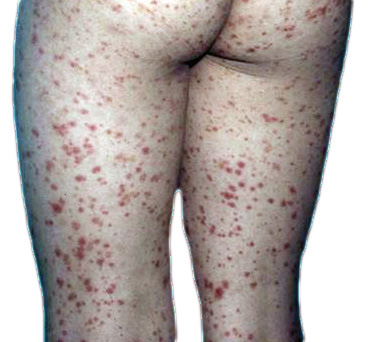
Figura 29. Lesiones eritemato purpúricas características de la púrpura de Schönlein Henoch.
- Urticarial: vasculitis, urticaria vasculitis hipocomplementémica, síndromes autoinflamatorios. Fiebre mediterránea familiar.
- Exantema poliformo: enfermedad de Kawasaki.
- Eritema periocular: lupus neonatal.
- Eritema periungueal: dermatomiositis juvenil.
- Eritema crónico migrans: enfermedad de Lyme.
- Eritema marginado: fiebre reumática.
• Raynaud (Fig. 30): enfermedad de Raynaud (idiopático), esclerosis sistémica, LES, vasculitis, síndrome de Sjögren y sindrome de desfiladero torácico (costilla cervical, etc.).
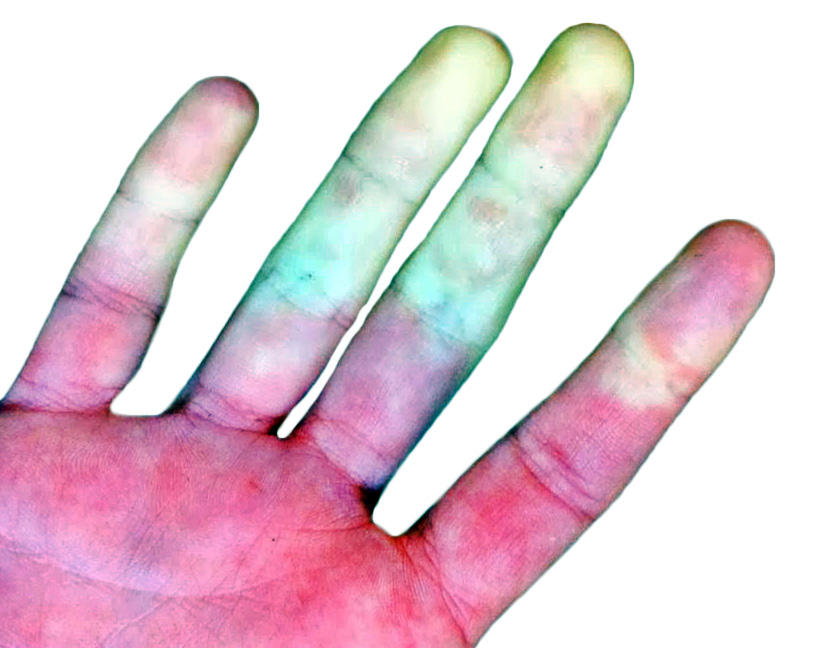
Figura 30. Fenómeno de Raynaud. Palidez cutánea que aparece en dedos de manos y pies (también, en otras zonas acras, como: nariz, pabellón auricular, areola mamaria, etc.) es consecuencia de episodios de vasoespasmos desencadenados por el frío o el estrés emocional. El cambio de coloración suele ser trifásico (palidez, cianosis y eritema). Por lo general, es benigno e idiopático y suele presentarse en mujeres jóvenes, pero puede indicar la presencia de una enfermedad autoinmune (LES, esclerodermia, EMTC, vasculitis, etc.) y, también, ser el primer síntoma de un síndrome de desfiladero torácico (costilla cervical), hipotiroidismo, diabetes, crioglobulinemia, etc.
• Livedo reticularis: vasculitis, síndrome de Snedon y síndrome antifósfolípido.
• Telangiectasias: síndrome de CREST y dermatomiositis juvenil (en párpados).
• Pápulas de Gottron (Fig. 31): Dermatomiositis juvenil.

Figura 31. Pápulas de coloración violácea o rosada en superficie extensora de dedos y rodillas (pápulas de Gottron), características de la dermatomiositis juvenil. Se aprecia exantema macular (signo de Gottron), como expresión de vasculitis.
• Calcificaciones subcutáneas: dermatomiositis juvenil, esclerodermia, enfermedad mixta del tejido conectivo y síndromes overlap.
• Púrpura palpable: vasculitis leucocitoclástica y vasculitis sistémica.
• Engrosamiento cutáneo: esclerodermia lineal / morfea (Fig. 32).
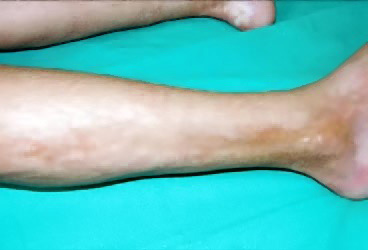
Figura 32. Esclerodermia localizada en pierna. Morfea.
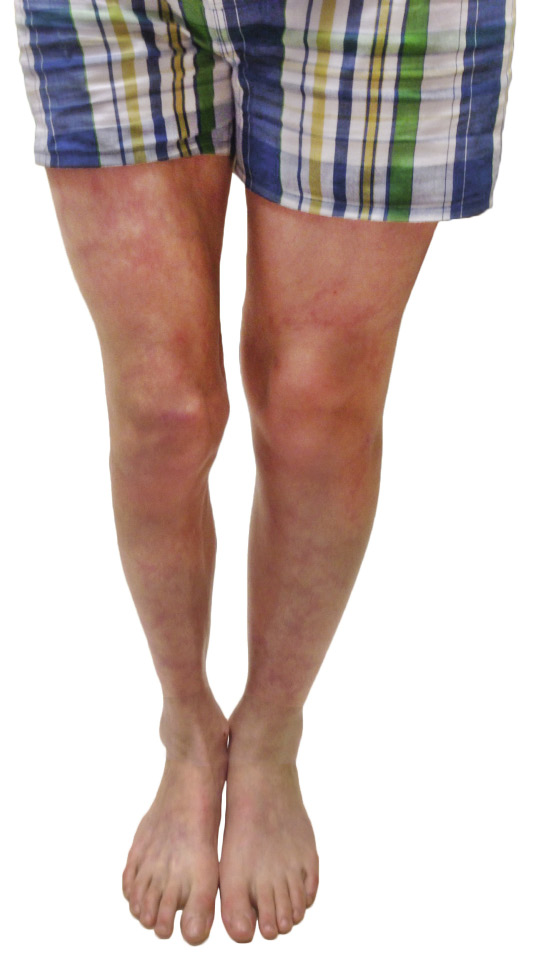
Figura 33. Atrofia importante MID en paciente con Morfea.
• Esclerodactilia: esclerodermia y síndromes relacionados.
• Nódulos subcutáneos: AIJ poliarticular y vasculitis tipo PAN.
• Paniculitis (eritema nodoso): sarcoidosis, enfermedad inflamatoria intestinal y vasculitis.
• Úlceras cutáneas: Vasculitis sistémicas.
• Úlceras digitales: LES y esclerodermia.
• Patología ungueal:
- Onicolisis: artritis psoriásica.
- Pitting: artritis psoriásica.
- Hemorragias en astilla: síndrome antifosfolipídico.
• Patergia: enfermedad de Behcet.
• Erisipela: fiebre mediterránea familiar.
Ocular
• Ojo rojo:
- Uveítis anterior crónica: AIJ, por lo general, es asintomática. Sarcoidosis.
- Uveítis anterior aguda (Fig. 34): espondiloartirtis juvenil y enfermedad de Kawasaki.
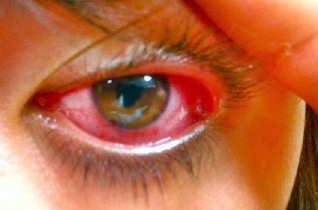
Figura 34. Hiperermia periciliar en paciente con uveítis aguda. Espondiloartritis juvenil.
- Conjuntivitis: síndrome de Reiter, enfermedad de Kawasaki, síndromes autoinflamatorios (criopirinopatías, TRAPS) y vasculitis.
- Epiescleritis: vasculitis sistémicas y enfermedades autoinflamatorias (criopirinopatías).
• Escleras azules: osteogénesis imperfecta.
• Miopía magna: síndrome de Stickler.
• Hipopion: enfermedad de Behcet.
ORL
• Faringo amigdalitis: síndrome PFAPA.
• Hipoacusia: síndromes autoinflamatorios (criopironopatías) y enfermedad inmunomediada del oído.
• Úvula bipartita: síndrome de Stickler.
• Condritis auricular: policondritis recidivante.
• Nariz en silla de montar: granulomatosis de Wegener.
• Aftas nasales: LES.
Cardiopulmonar
• Roce pleural: pleuritis lúpica y fiebre mediterránea familiar.
• Roce pericárdico: pericarditis lúpica, AIJ sistémica, fiebre mediterránea familiar y enfermedad autoinflamatoria (TRAPS).
• Arritmia: miocarditis lúpica y esclerosis sistémica.
• Soplos:
- Soplo insuficiencia aórtica: espondiloartropatías juveniles.
- Soplo mitral: prolapso mitral (hiperlaxitud generalizada).
- Soplo carotídeo: enfermedad de Takayasu.
• Disminución / ausencia murmullo: neumopatía intersticial reumatoide, hemorragia pulmonar (LES) y vasculitis.
• Sibilancias: vasculitis de Churg Straus.
Sistema nervioso
• Paresia pares craneales: LES. Enfermedad de Lyme. Vasculitis.
• Cefalea: LES. Vasculitis sistémicas.
• Convulsiones: LES y vasculitis sistémicas.
• Déficits focales. LES, síndromes autoinflamatorios y vasculitis.
• Corea: LES, síndrome antifosfolipídico y fiebre reumática.
• Meningitis aséptica: enfermedad de Behcet y síndromes autoinflamatorios.
• Mononeuritis múltiple: vasculitis sistémica.
La anamnesis y exploración física en Reumatología Pediátrica tiene sus peculiaridades y síntomas y signos guía que el médico de Atención Primaria, pediatra o reumatólogo deben conocer para orientar el diagnóstico y posterior abordaje de enfermedades musculoesqueléticas que, como el lector bien sabe, constituyen uno de los grupos de patología crónica más frecuente en la edad pediátrica.
Bibliografía
1. Rotes J. Exploración del aparato locomotor. En: Pascual E. Tratado de reumatología. Madrid: ARAN Editorial; 1998. p. 177-208.
2. Gill I, Sharif F. A disjointed effort: paediatric musculoskeletal examination. Arch Dis Child. 2012; 97: 641-3.
3. Goff I, Bateman B, Myers A, Foster H. Acceptability and practicality of musculoskeletal examination in acute general pediatric assessment. J Pediatr. 2010; 156: 657-62.
4. Clemente Garulo D, López Robledillo JC. Enfermedades reumáticas típicas o más frecuentes en la infancia. In: Enfermedades reumáticas de la infancia: aproximación diagnóstico-terapéutica. Guía práctica para el pediatra de atención primaria. Madrid, Publicis Healthcare communications Groups. 2012: 27-57.
5. López Robledillo JC. Enfoque práctico del dolor en niños y adolescentes. En: López Robledillo JC, Ed. Monografía SER de Reumatología pediátrica. Madrid: Editorial médica panamericana. 2007: 273-82.
6. López Robledillo JC. Síndrome del dolor musculoesquelético en la edad pediátrica. Pediatr Integral. 2013; XVII(1): 15-23.
7. Berard R. Approach to the child with joint inflammation. Pediatr Clin North Am. 2012; 59: 245-62.
8. Anthony KK, Schanberg LE. Pediatric pain syndromes and management of pain in children and adolescents with rheumatic disease. Pediatr Clin North Am. 2005; 52: 611-39.
9. Davies K, Woo P. Non-rheumatic causes of musculoskeletal symptoms in childhood (I). Acta Pediatr Esp. 2003; 61: 445-58.
10. Davies K, Woo P. Non-rheumatic causes of musculoskeletal symptoms in childhood (II). Acta Pediatr Esp. 2003; 61: 516-24.
11. Foster HE, Kay LJ, Friswell M, Coady D, Myers A. Musculoskeletal examination pGALS for school-age children based on the adult GALS screen. Arthritis Rheum. 2006; 55: 709-16.
































































































































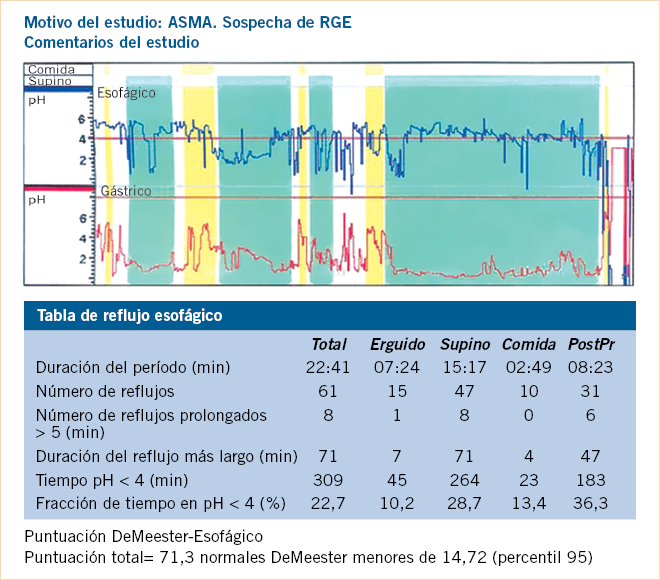




























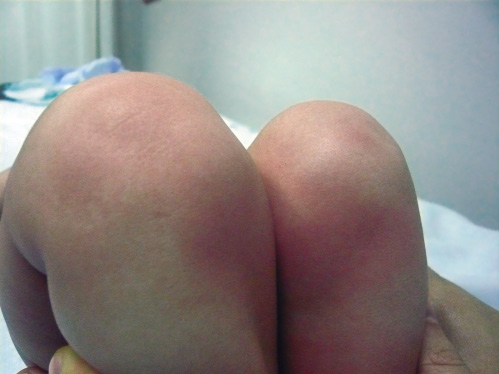

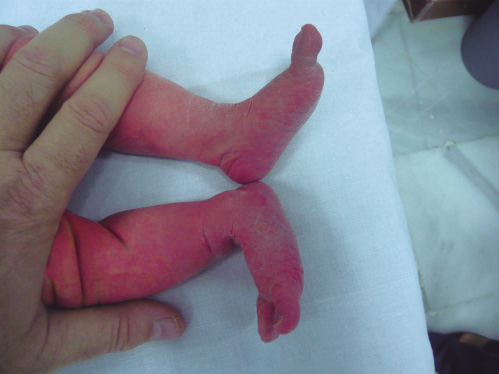
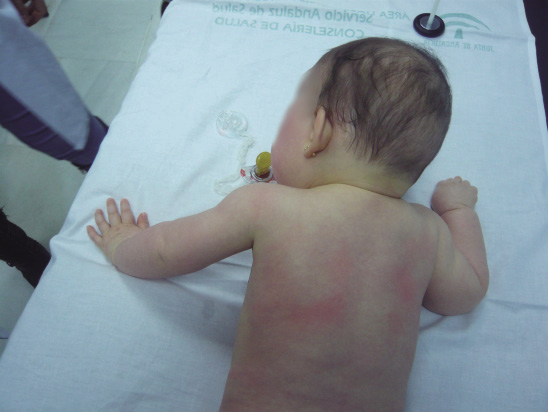
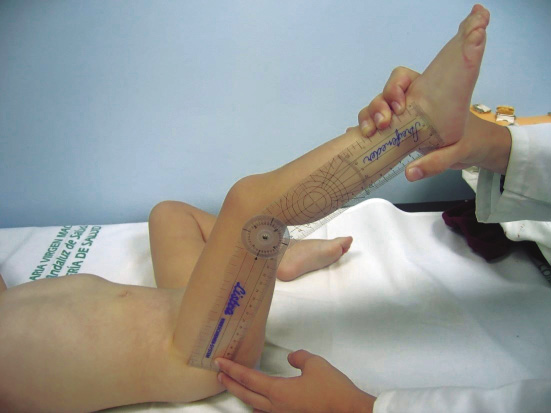





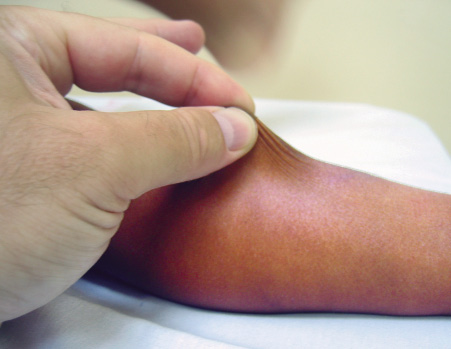
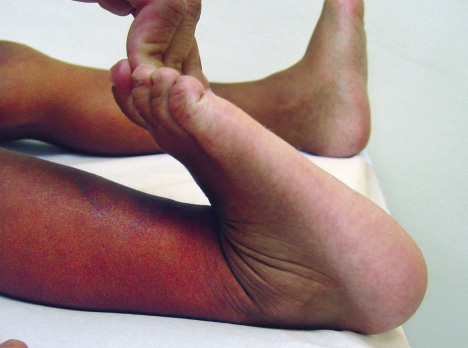










 Figura 1. Triángulo de Evaluación Pediátrica.
Figura 1. Triángulo de Evaluación Pediátrica.
