|
| Temas de FC |
B. Corredor Andrés*, M. Güemes**, M.T. Muñoz Calvo***
*Sección de Endocrinología, Hospital Virgen de la Salud. Toledo. **Servicio de Endocrinología, Hospital Infantil Universitario Niño Jesús. Madrid. ***Unidad de Endocrinología, Servicio de Pediatría. H. Ruber Internacional. Madrid
| Resumen
La hipercolesterolemia familiar (HF) es el trastorno genético más prevalente en la edad pediátrica; sin embargo, en la inmensa mayoría de los casos, pasa totalmente desapercibida. Se caracteriza por niveles plasmáticos elevados de C-LDL (lipoproteínas de baja densidad) que pueden detectarse ya desde el nacimiento, así como por presentar una alta tasa de morbimortalidad por enfermedad cardiovascular en edades tempranas. Se revisan las recomendaciones actuales de las guías de consenso en el tratamiento de la HF, describiéndose las características de la dieta preventiva y terapéutica, así como la promoción de la actividad física. Se recomienda iniciar tratamiento en la HF heterocigota entre los 8 y 10 años, tras 3-6 meses de dieta y ejercicio físico. Las estatinas deben ser incluidas entre los potenciales fármacos de primera línea, por la experiencia adquirida en los últimos años y por su capacidad de disminuir los niveles de C-LDL. Asimismo, se revisan las nuevas terapias emergentes. |
| Abstract
Although familial hypercholesterolemia (FH) is the most prevalent genetic disorder in the pediatric age, it remains underdiagnosed. It is characterized by high levels of plasma LDL-cholesterol (low density lipoproteins) since birth, and it associates a high morbidity and mortality rate due to cardiovascular disease (CVD) at an early age. The current treatment consensus recommendations are revised, including the characteristics of preventive and therapeutic nutrition as well as the promotion of physical activity. The treatment onset for heterozygous FH is recommended between the ages of 8 and 10 years, following 3-6 months of diet and physical exercise. Statins should be included among the potential first-line agents given the experience attained over the last years and considering their ability to decrease LDL- cholesterol concentrations. In addition, new emerging therapies are reviewed. |
Palabras clave: Hipercolesterolemia; Colesterol total; LDL-colesterol; Cribado; Estatinas.
Key words: Hypercholesterolemia; Total cholesterol; LDL-cholesterol; Screening; Statins.
Pediatr Integral 2020; XXIV (3): 166 – 173
Hipercolesterolemia familiar en la infancia y la adolescencia: cribado, diagnóstico y tratamiento
Introducción
La hipercolesterolemia familiar (HF) es una enfermedad genética monogénica, caracterizada por niveles plasmáticos elevados de C-LDL (lipoproteínas de baja densidad) que pueden detectarse ya desde el nacimiento, así como por presentar una alta tasa de morbimortalidad por enfermedad cardiovascular (ECV) en edades tempranas.
Existen dos formas de HF, la forma heterocigota (HFHe) es el trastorno hereditario más frecuente, con una prevalencia de 1/200-250 individuos, y la forma homocigota (HFHo), con una prevalencia de 1/400.000 individuos(1). En menores de 18 años en nuestro país, la prevalencia con fenotipo de HFHe es de 1/217 individuos(2). En países con programas de cribado genético como Holanda, se alcanzan porcentajes de detección superiores al 70%. Los adolescentes con HFHo presentan un elevado riesgo, desarrollando enfermedad coronaria antes de los 20 años si no son tratados intensamente, y generalmente fallecen antes de los 30 años. Por ello, el diagnóstico y el tratamiento precoz son muy importantes para su pronóstico y evolución a largo plazo(3).
Hay evidencias que demuestran que los niveles elevados de C-LDL en el niño, inducen a la formación y al desarrollo de la lesión ateromatosa en edades tempranas. El grosor íntima-media de la carótida de niños con fenotipo de HF es superior si se compara con el de niños normolipémicos, estando relacionado directamente con los niveles de C-LDL. Esta diferencia fue observada en niños a partir de los siete años(4).
La detección de la HF es un gran desafío para el pediatra, ya que la inmensa mayoría no son diagnosticados, lo que conlleva un retraso en el inicio del tratamiento, (cambios en los estilos de vida y farmacológico), y este hecho puede contribuir a elevar el riesgo cardiovascular de esta población en la edad media de vida(1).
Niveles de lipoproteínas en la infancia y la adolescencia
Los niveles plasmáticos de lipoproteínas son diferentes en la infancia y la adolescencia en comparación con la edad adulta.
Los niveles de colesterol total (CT), C-LDL, colesterol unido a proteínas de alta densidad (C-HDL) y triglicéridos (TG) ascienden paulatinamente desde el nacimiento y se estabilizan entre los 2 y 4 años, manteniéndose en un mismo percentil a lo largo del tiempo durante los años prepuberales (fenómeno tracking del colesterol). A partir de los 10-12 años, los niveles plasmáticos de CT y C-LDL disminuyen entre un 5 y un 10% en ambos sexos –siendo más evidente en varones–, debido al descenso acusado del C-HDL, pero en los últimos años de la adolescencia, se produce un nuevo ascenso de los niveles de CT y C-LDL alcanzando niveles medios de adulto a partir de los 20 años. La disminución de los niveles de C-HDL en varones es el patrón más aterogénico que se produce durante la pubertad, y va a permanecer durante la etapa adulta(5).
Para evaluar el riesgo de ECV en función de los niveles de CT, C-LDL, C-HDL y triglicéridos, la Guía del National Heart, Lung, and Blood Institute (NHLBI), National Institutes of Health de EE.UU. ha propuesto unos puntos de corte de los lípidos plasmáticos (Tabla I). Existen diferencias en las concentraciones de colesterol en función de la edad, sexo y desarrollo puberal(5-6).
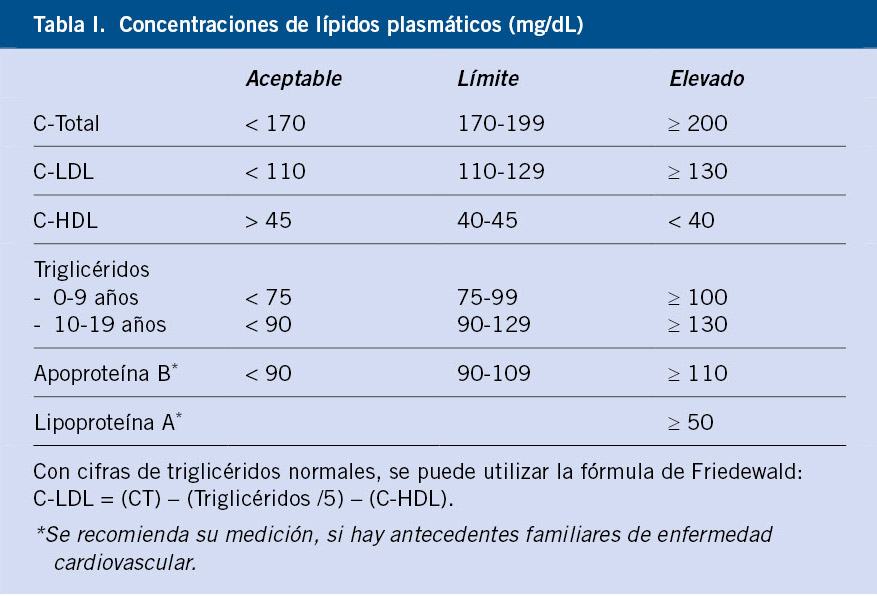
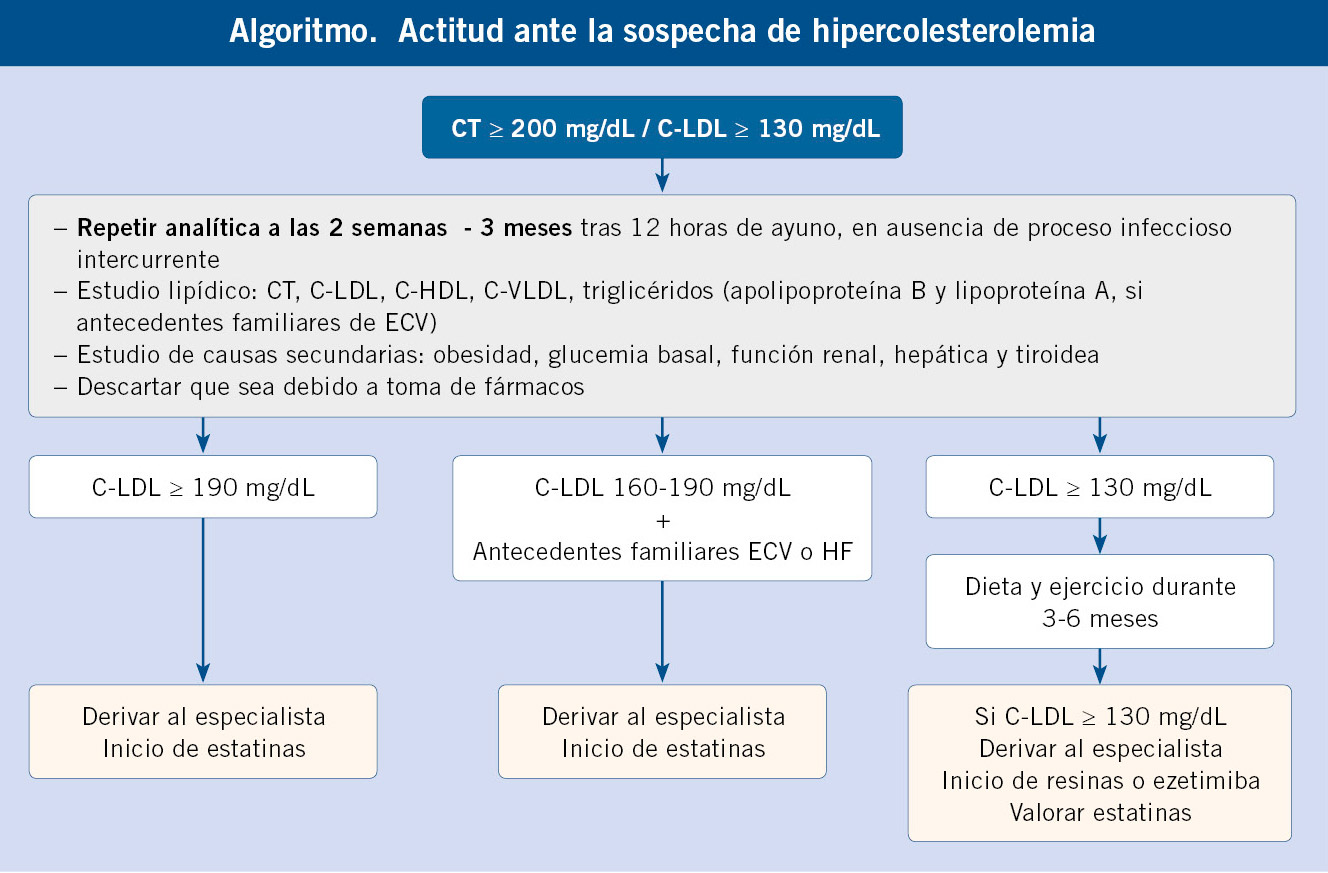
De modo práctico, debe considerarse como hipercolesterolemia, concentraciones de CT y C-LDL superiores al percentil 95: CT ≥ 200 mg/dL y C-LDL ≥ 130 mg/dL. Además, deben considerarse anormales niveles de triglicéridos > 100-130 mg/dL y los niveles de colesterol unido a lipoproteínas de alta densidad (C-HDL) < 40 mg/dL(6–8).
Identificación de la población pediátrica de riesgo
Existen distintas estrategias para el diagnóstico (Fig. 1):

Figura 1. Cribado de hipercolesterolemia en pediatría. IMC: índice de masa corporal; FC: frecuencia cardiaca; TA: tensión arterial; HF: hipercolesterolemia familiar; DE: desviación estándar; ECV: enfermedad cardiovascular; ACV: accidente cerebro vascular.
• Cribado universal: consiste en la determinación de los niveles de CT de forma rutinaria, a los niños en una edad concreta. Se ha demostrado que este método permite detectar el 90% de los niños con HF(9).
• Cribado selectivo: consiste en la determinación de los niveles de CT en todos aquellos con historia familiar de ECV o con historia familiar de hipercolesterolemia en alguno de los progenitores. Este tipo de cribado ha sido recomendado por el Programa Nacional de Colesterol de EE.UU., la Asociación Americana del Corazón y la Academia Americana de Pediatría, detectándose solo el 30-60% de niños con HF(9).
• Cribado en cascada directa: si conocemos la variante genética patológica causal de la HF del progenitor, se extiende el estudio genético a los familiares de primer grado, incluyendo a los niños. Este tipo de cribado genético tiene el 100% de sensibilidad y especificidad en el estudio de los familiares, recomendándose por ser la mejor técnica por su coste-efectividad(10).
• Cribado en cascada inversa: a partir de la detección de hipercolesterolemia en el niño, se inicia el estudio de los progenitores. Si uno de estos presenta una puntuación ≥ 6 en los criterios clínicos del “Dutch Lipid Clinic Network”(12), se solicitará el estudio genético(11).
• Cribado oportunista: la determinación de CT en cualquier estudio analítico(11).
Diagnóstico
No hay un criterio único respecto a la edad en la que se debe hacer el diagnóstico de hipercolesterolemia. En general, se recomienda entre los 2 y los 10 años. Su importancia radica en que cuanto antes se realice, más fácil será la adherencia a los hábitos de vida saludables.
La sospecha diagnóstica deberá establecerse en función de los niveles elevados de C-LDL y la historia familiar de hipercolesterolemia y/o de ECV(12).
• Si la concentración de CT es ≥ 200 mg/dL o C-LDL ≥130 mg/dL, se debe repetir el análisis tras 3 meses en condiciones estandarizadas (dieta normal, tras 12 horas de ayuno) con determinación de C-HDL, C-LDL, triglicéridos, y descartar las causas secundarias de hipercolesterolemia. Si existen antecedentes familiares de ECV, se recomienda la medición de: apoproteína B (ApoB), apoproteína A (ApoA) y lipoproteína A (Lpa). Niveles superiores a 30-50 mg/dL de Lpa son un buen factor predictor de ECV, y el índice ApoB/ApoA1 > 0,82 se ha considerado buen marcador para la detección de niños con HF(2,12) (V. algoritmo).
• Las causas más frecuentes de hipercolesterolemias secundarias son: hipotiroidismo, consumo de alcohol, ciertos medicamentos (contraceptivos, corticoides, beta bloqueantes, retinoides, antirretrovirales, anabolizantes, etc.). La hipercolesterolemia secundaria a la obesidad se caracteriza por hipertrigliceridemia y niveles disminuidos de C-HDL. Otras enfermedades que cursan con hipercolesterolemia suelen dar sintomatología evidente de la enfermedad primaria: diabetes, hepatopatías, síndrome nefrótico, enfermedades de depósito, etc.(3,13).
• Debemos recomendar un periodo de dieta de tres a seis meses. Si tras este periodo, en una nueva determinación, persisten los niveles de C-LDL ≥130 mg/dL, debemos sospechar una HF(5).
• Si los niveles de C-LDL son ≥190 mg/dL, obtenidos en dos determinaciones consecutivas con un intervalo de 2-3 meses, las probabilidades de hallar una mutación causal de HF son muy elevadas. Si presenta niveles de C-LDL ≥ 160 mg/dL tras un periodo de tratamiento dietético y/o existe una historia de ECV en familiares de primer grado (hombres <55 años, mujeres <60 años), indicaría una alta probabilidad de ser portador de una mutación causal de HF (v. algoritmo).
Es recomendable realizar el estudio genético ante la sospecha de hipercolesterolemia familiar. Se debe efectuar en un laboratorio acreditado y debe incluir la secuenciación completa para identificar mutaciones puntuales y deleciones/inserciones para los genes: RLDL, APOB y PCSK9 (13).
Se debe recordar que la ausencia de detección de una mutación no descarta el diagnóstico; debido a que, un 5-30% de los casos son debidos a causa genética no identificada.
Hipercolesterolemia familiar heterocigota (HFHe)
• El mecanismo de transmisión es autosómico dominante y, aproximadamente, la mitad de la descendencia de una persona afectada presentará la enfermedad. Se produce principalmente por mutaciones en el gen del receptor LDL (RLDL). Actualmente, se han descrito más de 1.700 mutaciones diferentes de este gen, situado en el cromosoma 19 (p13.2). En menor proporción, se han descrito defectos en el gen que codifica la Apo B y en el gen que codifica la proproteína convertasa subtilisina-kesina tipo 9 (PCSK9), representando entre el 5 y 1%, respectivamente(10).
• El diagnóstico clínico se basará en: concentraciones elevadas de C-LDL (entre 190-500 mg/dL), historia familiar de hipercolesterolemia, antecedentes de ECV y presencia de arco corneal y/o xantomas (Fig. 2).

Figura 2. A. Arco corneal juvenil, línea blanco-grisácea situada en los márgenes de la córnea, debido a depósito extracelular de partículas lipídicas, principalmente ésteres de colesterol, dentro del estroma corneal en la región del limbo esclero-corneal.
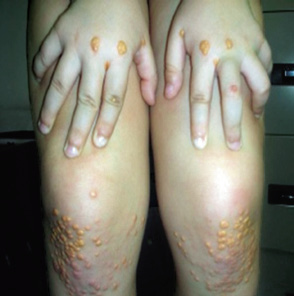
Figura 2. B.Xantomas: acúmulo de macrófagos espumosos en tejido conectivo dérmico y tendones, donde se producen masas tumorales.
• La prevalencia y el elevado riesgo de desarrollar ECV hacen de la HFHe un problema de salud pública y, aun así, la mayoría de los pacientes están sin diagnosticar ni tratar. El diagnóstico precoz permite adecuar medidas preventivas que reducirían el riesgo de ECV(1,3,10,13).
Hipercolesterolemia familiar homocigota (HFHo)
Es una enfermedad rara y potencialmente mortal que se caracteriza clínicamente por la presencia de: niveles plasmáticos de CT (superior a 500 mg/dL), xantomas antes de los 10 años y ECV prematura (Tabla II).

Es debida a un defecto de la capacidad de unión e internalización de las partículas de LDL, generalmente causado por mutaciones en los dos alelos del RLDL (>95%), y cada uno de sus progenitores presenta una HFHe. Recientemente, se han identificado mutaciones en alelos de otros tres genes secundarios: APOB (2-5%), PCSK9 (<1%) y LDLRAP1 (<1%, que codifica la proteína adaptadora 1 del receptor de LDL). Los pacientes son homocigotos, con la misma mutación en ambos alelos del mismo gen o, más a menudo, heterocigotos compuestos con mutaciones diferentes en cada alelo del mismo gen, o heterocigotos dobles con mutaciones en dos genes diferentes que afectan a la función del receptor de LDL(14). Independientemente del defecto genético, la gravedad del fenotipo de HFHo depende de la actividad residual del receptor de LDL (LDLR). Según los ensayos in vitro realizados con los fibroblastos cultivados, se han clasificado como pacientes con receptor negativo (actividad residual < 2%) o con receptor defectuoso (actividad residual 2-25%). Los pacientes con HFHo que son LDLR negativo tienen niveles más altos de C-LDL y un pronóstico clínico más desfavorable que los pacientes con LDLR defectuoso. Si no reciben tratamiento, la mayoría de estos pacientes desarrollan arteriosclerosis sintomática antes de los 20 años y generalmente fallecen antes de los 30 años(14).
Diagnóstico diferencial
• Hipercolesterolemia poligénica: es la más frecuente de las hipercolesterolemias primarias y la de riesgo menos elevado. La prevalencia en población general es aproximadamente del 4%. Se caracteriza por niveles de C-LDL discretamente elevados, mayor de 130 mg/dL, con padres y hermanos con concentraciones similares (agregación familiar).
Su etiología es desconocida, se piensa que ocurre por alteraciones en diversos genes reguladores interaccionando con factores ambientales, especialmente la dieta, que darían lugar al aumento del colesterol total. Suele presentarse a partir de la segunda década de la vida(3).
• Hipercolesterolemia familiar combinada: es genéticamente heterogénea, solo el 10-20% muestran niveles elevados en la infancia (usualmente, en forma de hipertrigliceridemia). La prevalencia es del 1-2% en la población general. Su diagnóstico requiere niveles de C-LDL y/o triglicéridos en percentil mayor de 95 (aproximadamente mayor de 130-140 mg/dL y mayor de 110 mg/dL, respectivamente). Las lipoproteínas de uno de los progenitores deben tener igual comportamiento (C-LDL y/o triglicéridos superiores a 160 y 200 mg/dL, respectivamente). Suele presentarse a partir de la segunda década de la vida y es frecuente la asociación con obesidad, hipertensión arterial y diabetes tipo 2(5).
• Sitosterolemia: tiene un patrón de herencia autosómica recesiva, por lo que los progenitores pueden presentar niveles normales de colesterol. Presentan: concentraciones plasmáticas marcadamente elevadas (>30 veces) de fitoesteroles, niveles elevados de colesterol que responden bien a la dieta y a los secuestradores de ácidos biliares o ezetimiba, y xantomas tendinosos y/o tuberosos que pueden desaparecer después de las dos primeras décadas de vida. El diagnóstico se confirma mediante análisis genético, por mutación del transportador ABCG5/ABCG8 implicado en el transporte del colesterol no esterificado por las células epiteliales intestinales hacia el lumen del intestino(3).
• Hipercolesterolemia autosómica recesiva: es debida al déficit del adaptador de la proteína ARH o a la incapacidad del mismo para interactuar con el receptor de LDL, por ello cursa con disminución del aclaramiento de LDL y aumento de C-LDL y CT. Su fenotipo es similar a la hipercolesterolemia familiar homocigota pero, en general, menos severo y con mayor respuesta a los fármacos hipolipemiantes. Cursa con xantomas grandes desde la infancia, niveles de CT mayor de 500 mg/dl, y aparición de ECV en menores de 30 años(3).
• Disbetalipoproteinemia: tiene un patrón de herencia autosómico recesivo, y es secundaria a alteraciones en el gen APOE, localizado en el cromosoma 19 (19q 13.2), con predominio de la isoforma Apo E2. Su incidencia en la población general es de un caso por 10.000. Cursan con hipercolesterolemia e hipertrigliceridemia y, en algunos casos, con xantomas estriados palmares(3,5,12).
Tratamiento
Debe ser multidisciplinar y comprender: una dieta equilibrada, un ejercicio físico regular y un tratamiento farmacológico(12).
Dietético
El objetivo primordial del tratamiento dietético de la hipercolesterolemia será lograr que los niveles de C-LDL disminuyan, consiguiendo un descenso de un 10 a un 15%, aunque existen grandes variaciones individuales y dependiendo del tipo de mutación.
La ingesta calórica debe ser adecuada para favorecer el crecimiento y desarrollo de los niños y adolescentes, recomendándose:
• Hidratos de carbono: 50-60%; proteínas: 15%; y grasas: 25-30%; de las calorías totales.
• Ácidos grasos saturados <8-10% de las calorías totales.
• Ácidos grasos monoinsaturados + ácidos grasos poliinsaturados ≤ 20% del total de calorías.
• Colesterol máximo diario de 300 mg, aunque quizás sería más aconsejable la recomendación de 100 mg/1.000 calorías.
• Restricción de ácidos grasos con isómeros trans (<1% de calorías).
• Fibra dietética soluble de 5 a 15 g al día. Se ha demostrado que el uso de alimentos ricos en fibra soluble, como el salvado de avena, reducen los niveles de TG y C-LDL. Es recomendable el aumento de consumo de fibra de forma progresiva, para evitar posibles molestias intestinales(12).
• Uso de esteroles y estanoles vegetales 1,5-2 g/día en niños mayores de 6 años con HFHe. Se ha visto que podrían reducir los niveles de C-LDL entre un 8-10%, aunque son necesarios ensayos clínicos que investiguen su efecto en la reducción de eventos cardiovasculares(12).
• Micronutrientes: vigilar especialmente el aporte de vitaminas liposolubles (especialmente vitamina D) y minerales (hierro, zinc y calcio)(13).
Actividad física
Los niños y adolescentes deben dedicar un número establecido de horas al día al ejercicio físico en los colegios, y se debe promover su participación en actividades deportivas. Se debe preguntar sobre sus intereses y los medios disponibles a su alcance, para poder realizar un plan personalizado. De la misma manera, se deben potenciar las actividades deportivas en familia(3).
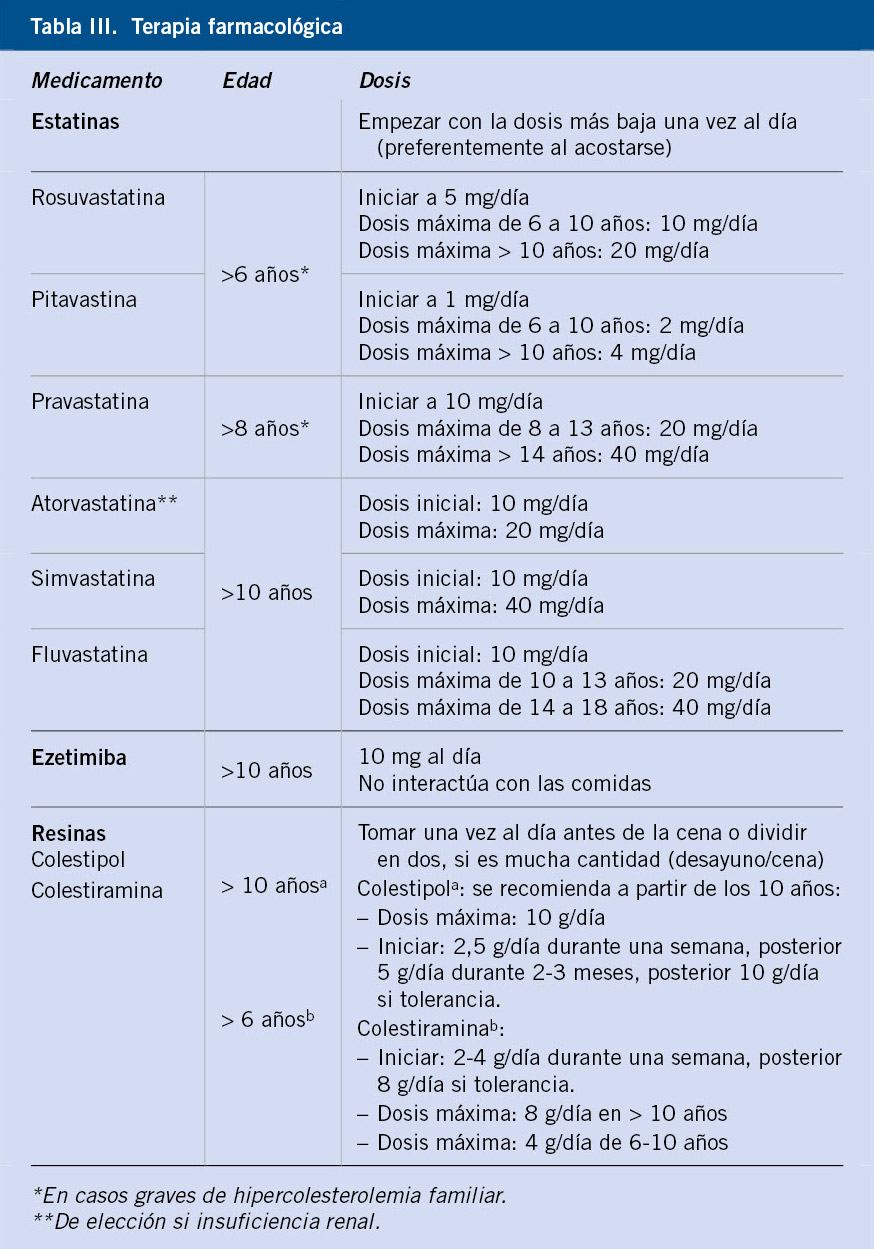
Farmacológico (Tabla III)
Estatinas
Deben ser incluidas entre los potenciales fármacos de primera línea, por la experiencia adquirida en los últimos años y por su capacidad de disminuir los niveles de C-LDL en torno a 18-45%, y sin afectación del crecimiento ni del desarrollo puberal(15).
Los objetivos del tratamiento recomendados en la HFHe son: en niños ≥10 años, llegar a niveles de C-LDL <130 mg/dl, y en niños entre 8 y 10 años una reducción del 30-50% respecto a los niveles previos de C-LDL.
Edad de inicio
Diferentes guías recomiendan iniciar tratamiento en la HFHe, entre los 8 y 10 años tras 3-6 meses de dieta y ejercicio físico(15).
Se recomienda iniciar tratamiento si los niveles de:
• C-LDL ≥ 190 mg/dl.
• C-LDL ≥ 160 mg/dl con historia de ECV prematura en familiares de primer grado y/o si existen otros factores de riesgo cardiovascular (tabaco, C-HDL bajo, lipoproteína A elevada, hipercolesterolemia familiar) o presencia de enfermedades con elevado riesgo cardiovascular (diabetes, síndrome metabólico, obesidad, hipertensión arterial, enfermedades inflamatorias crónicas, VIH, insuficiencia renal crónica, trasplante de órganos).
En la actualidad, están aprobados, por la FDA y la Agencia Europea del Medicamento (EMA):
• Rosuvastatina y Pitavastina, a partir de los 6 años.
• Pravastatina, a partir de los 8 años.
• Atorvastatina, Lovastatina, Simvastatina y Fluvastatina, en mayores de 10 años.
Dosificación
• Empezar con la dosis más baja una vez al día, generalmente al acostarse. Medir niveles basales de CPK, ALT y AST(12).
• Tras 4 semanas de tratamiento, determinar: perfil de lipoproteínas en ayunas, CPK, ALT y AST. Repetir el control analítico a las 8 semanas y a los 3 meses. Posteriormente, cada 3-6 meses en función de los resultados.
• Si hay anomalías de laboratorio (elevación de transaminasas tres veces superior al límite de la normalidad) o aparecen síntomas, suspender temporalmente el fármaco y repetir analítica en 2 semanas. Cuando los valores retornen a la normalidad, el fármaco puede reiniciarse con monitorización estrecha(12).
• Para evitar elevaciones de CPK secundarias a ejercicio físico intenso, se deberá evitar realizar ejercicio físico intenso tres días antes de la extracción sanguínea.
Seguimiento
• Valorar el ritmo de crecimiento y el desarrollo puberal (peso, talla, índice de masa corporal, estadio de Tanner).
• Monitorizar el perfil de lipoproteínas en ayunas, CPK, ALT y AST cada 3-6 meses.
• Insistir al paciente sobre medidas dietéticas e informar sobre otros factores de riesgo, tales como: sobrepeso, sedentarismo y tabaquismo, entre otros.
Eficacia y seguridad
• Se han publicado revisiones sobre su utilización en la infancia, concluyendo que, a la vez de seguras son muy eficaces. Un estudio de seguimiento en 214 niños con HF tratados con Pravastatina durante 20 años, ha demostrado su seguridad a largo plazo y la reducción del engrosamiento de la íntima-media de la carótida(15,16).
• En cuanto a los efectos adversos, no se han publicado casos de miositis, miopatía o rabdomiólisis. Se han encontrado algunos casos de elevaciones asintomáticas de la CPK y enzimas hepáticas, que han revertido con un descenso de las dosis de la medicación sin necesidad de suspender el tratamiento. Tampoco se han encontrado alteraciones del ritmo de crecimiento, desarrollo puberal ni del metabolismo de las vitaminas.
• Las estatinas están contraindicadas en el embarazo, por lo que debe advertirse a las adolescentes y remitirlas, en caso necesario, a consejo ginecológico.
• Si el niño presenta insuficiencia renal, estaría indicada la atorvastatina, ya que su eliminación renal es mínima(12,15,16).
Ezetimiba
Actúa inhibiendo selectivamente la absorción intestinal de colesterol (tanto dietético como de origen biliar) en el borde en cepillo de los enterocitos.
Es un fármaco de segunda línea y se puede administrar de forma combinada con una estatina. Los principales efectos adversos son: gastrointestinales (diarrea y dolor abdominal) y cefalea. Reducen los niveles de C-LDL un 20%.
Su uso está autorizado por la FDA y la EMA, a partir de los 10 años de edad. No existen datos de seguridad a largo plazo en población pediátrica(17).
Resinas de intercambio iónico
Actúan inhibiendo la absorción de ácidos biliares a nivel intestinal. No se absorben y son seguras a largo plazo. Sin embargo, por su baja palatabilidad y por sus efectos adversos a nivel gastrointestinal como flatulencia y estreñimiento, han caído en desuso. Reducen los niveles de C-LDL de un 16% a un 19%(12).
Indicado en niños mayores de 6 años con hipercolesterolemia grave en los que aún no se consideren indicadas las estatinas. La dosis media recomendada es de 0,25-0,35 g/kg/día, con una dosis máxima de 10 g/día para el colestipol y de 8 g para la colestiramina. Se debe iniciar el tratamiento con dosis bajas, e incrementar progresivamente.
En tratamientos prolongados, se puede alterar la absorción de vitaminas liposolubles y de ácido fólico, por lo que puede ser necesaria su monitorización. El Colesevelam es la resina comercializada más recientemente, y ha sido autorizado por la FDA y no por la EMA. Puede administrase en monoterapia o en combinación con estatinas en niños mayores de 10 años(18).
Nuevas moléculas hipolipemiantes
Inhibidores PCSK9
La PCSK9 es una proteína enzimática de la familia de la subtilisina de serinas-proteasas, que se sintetiza primariamente en el hígado, aunque también se encuentra en el intestino y riñón. Son un nuevo grupo de fármacos, con gran eficacia y seguridad.
La inhibición de PCSK9 reduce el número de receptores que van a ser degradados, aumentando así su densidad en la superficie celular, con la subsiguiente reducción del colesterol plasmático. En España, han sido aprobados evolocumab y alirocumab en HFHe en población adulta, en la que con una dosis máxima tolerada de hipolipemiantes no se consigue alcanzar los objetivos terapéuticos.
Niños con HFHo también han sido incluidos en los ensayos clínicos con evolocumab, pero no se ha establecido la seguridad en menores de 12 años. Se han observado reducciones del C-LDL en niños portadores de receptores LDL defectuosos; sin embargo, no se observó ningún efecto en los portadores de receptores nulos. Esto ayudaría a disminuir las sesiones de LDL-aféresis(13,19).
Lomitapida
Esta molécula de administración oral, inhibe la proteína de transferencia microsomal de triglicéridos, una enzima clave en el acoplamiento y la secreción de lipoproteínas que contienen ApoB, tanto en el hígado (VLDL con ApoB 100) como en el intestino (quilomicrones con ApoB 48). Por tanto, la inhibición de esta enzima disminuye la síntesis y secreción de la VLDL y de los quilomicrones, y así es capaz de reducir los niveles plasmáticos de CT, TG, C-LDL y C-VLDL(3).
Ha sido aprobada por la FDA y la EMA para el tratamiento de la HFHo. Se ha demostrado en un estudio, que puede reducir hasta en un 50% adicional la concentración de C-LDL. Los efectos secundarios son muy frecuentes y fundamentalmente son de tipo gastrointestinal. Asimismo, se han descrito elevaciones de las transaminasas y desarrollo de esteatosis hepática; ello exigirá su monitorización a lo largo del tratamiento(13).
Mipomersen
Es un oligonucleótido antisentido que inhibe la transcripción del ARNm de la ApoB. La reducción de la síntesis de la ApoB da lugar a una disminución de las VLDL a nivel intrahepático y, consecuentemente, del C-LDL. Recientemente, se ha publicado un estudio a largo plazo, donde el tratamiento con este fármaco se asocia a una reducción de eventos CV en pacientes con HF(13).
No ha sido aprobado por la EMA, debido a la presencia de numerosos efectos adversos, fundamentalmente reacciones locales en el lugar de la inyección y elevación de las transaminasas(12,13).
Tratamiento de la HFHo
• Se basa en una combinación de cambios del estilo de vida, estatinas con ezetimiba y aféresis de LDL.
• La terapia hipolipemiante con fármacos debe iniciarse a partir de los 2 años.
• La aféresis de LDL se debe realizar en todos los pacientes y debe iniciarse preferentemente a los 5 años y no más tarde de los 8 años.
• Lomitapida debería considerarse como tratamiento complementario, para reducir adicionalmente los niveles plasmáticos de C-LDL en pacientes con HFHo, con o sin aféresis.
• Los métodos hormonales anticonceptivos están contraindicados en la HFHo. Las mujeres que deseen quedarse embarazadas deberán recibir asesoramiento y someterse a una evaluación cardiovascular. Las mujeres embarazadas deberán tratarse con aféresis de LDL.
• El apoyo psicológico debería integrarse dentro de la atención médica habitual.
• Los grupos de apoyo al paciente y a la familia tienen un papel relevante.
• Puede considerarse la cirugía para extirpar xantomas cutáneos y/o tendinosos grandes por motivos funcionales o estéticos(14).
LDL aféresis
Es una estrategia costo-efectiva y segura. Se recomienda iniciarla antes de los 5 años y nunca después de los 8 años. La reducción de C-LDL y lipoproteína A conseguida se encuentra entre un 50 y un 75% de los niveles basales, cuando se utiliza de forma semanal o cada dos semanas.
El tratamiento con estatinas se debe mantener para retrasar el efecto rebote en el aumento de los niveles de C-LDL. Es un procedimiento bien tolerado, con efectos adversos en menos del 5%, que incluyen: hipotensión, dolor abdominal, náuseas, hipocalcemia, anemia ferropénica y aquellos relacionados con el acceso venoso(3,14,20).
Bibliografía
Los asteriscos muestran el interés del artículo a juicio del autor.
1. Wilemon KA, Patel J, Aguilar-Salinas C, Ahmed CD, Alkhnifsawi M, Almahmeed W, et al. Reducing the Clinical and Public Health Burden of Familial Hypercholesterolemia: A Global Call to Action. JAMA Cardiol. 2020. doi:10.1001/jamacardio.2019.51732.
2. Plana N, Rodríguez-Borjabad C, Ibarretxe D, Ferré R, Feliu A, Caselles A, et al. Lipid and lipoprotein parameters for detection of familial hypercholesterolemia in childhood. The DECOPIN Project. Clin Investig Arterioscler. 2018; 30: 170-8.
3.*** Mata P, Alonso R, Ruiz A, González-Juanatey JR, Badimón L, Díaz-Díaz JL, et al. Diagnóstico y tratamiento de la hipercolesterolemia familiar en España: documento de consenso. Semergen. 2015; 41: 24-33.
4. Wiegman A, De Groot E, Hutten BA, Rodenburg J, Gort J, Bakker HD, et al. Arterial intima-media thickness in children heterozygous for familial hypercholesterolaemia. Lancet. 2004; 363: 369-70.
5. Muñoz Calvo MT. Dislipemias. Pediatr Integr. 2015; XIX(5): 355-64.
6. American Academy of Pediatrics. National Cholesterol Education Program: Report of the Expert Panel on Blood Cholesterol Levels in Children and Adolescents. Pediatrics. 1992; 89: 525-84.
7. Grundy SM, Stone NJ, Bailey AL, Beam C, Birtcher KK, Blumenthal RS, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Vol. 139, Circulation. Lippincott Williams and Wilkins; 2019. p. E1082-143.
8. Balder JW, Lansberg PJ, Hof MH, Wiegman A, Hutten BA, Kuivenhoven JA. Pediatric lipid reference values in the general population: The Dutch lifelines cohort study. J Clin Lipidol. 2018; 12: 1208-16.
9. Lázaro P, Pérez de Isla L, Watts GF, Alonso R, Norman R, Muñiz O, et al. Cost-effectiveness of a cascade screening program for the early detection of familial hypercholesterolemia. J Clin Lipidol. 2017; 11: 260-71.
10.*** Wiegman A, Gidding SS, Watts GF, Chapman MJ, Ginsberg HN, Cuchel M, et al. Familial hypercholesterolæmia in children and adolescents: Gaining decades of life by optimizing detection and treatment. Vol. 36, European Heart Journal. Oxford University Press; 2015. p. 2425-37.
11.*** Kusters DM, de Beaufort C, Widhalm K, Guardamagna O, Bratina N, Ose L, et al. Paediatric screening for hypercholesterolaemia in Europe. Arch Dis Child. 2012; 97: 272-6.
12.*** Mach F, Baigent C, Catapano AL, Koskina KC, Casula M, Badimon L, et al. 2019 ESC/EAS guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Atherosclerosis. 2019; 290: 140-205.
13.*** Plana N, Rodríguez-Borjabad C, Ibarretxe D, Masana L. Hipercolesterolemia familiar en la infancia y la adolescencia: una realidad oculta. Clin Investig Arterioscler. 2107; 29: 129-40.
14. Ascaso JF, Mata P, Arbona C, Civeira F, Valdivielso P, Masana L. Hipercolesterolemia familiar homocigota: Adaptación a España del documento de posición del grupo de consenso sobre hipercolesterolemia familiar de la Sociedad Europea de Arteriosclerosis. Documento de Consenso de la Sociedad Española de Arteriosclerosis (SEA) y la Fundación Hipercolesterolemia Famil. Clin Investig Arterioscler. 2015; 27: 80-96.
15. Luirink IK, Wiegman A, Kusters DM, Hof MH, Groothoff JW, De Groot E, et al. 20-Year follow-up of statins in children with familial hypercholesterolemia. N Engl J Med. 2019; 381: 1547-56.
16. Vuorio A, Kuoppala J, Kovanen PT, Humphries SE, Tonstad S, Wiegman A, et al. aStatins for children with familial hypercholesterolemia. Vol. 2019, Cochrane Database of Systematic Reviews. John Wiley and Sons Ltd; 2019.
17. Kusters DM, Caceres M, Coll M, Cuffie C, Gagné C, Jacobson MS, et al. Efficacy and safety of ezetimibe monotherapy in children with heterozygous familial or nonfamilial hypercholesterolemia. J Pediatr. 2015; 166: 1377-84.e3.
18. Davidson M. The efficacy of colesevelam HCl in the treatment of heterozygous familial hypercholesterolemia in pediatric and adult patients. Clin Ther. 2013; 35: 1247-52.
19. Ogura M. PCSK9 inhibition in the management of familial hypercholesterolemia. J Cardiol. 2018; 71: 1-7.
20. Luirink IK, Determeijer J, Hutten BA, Wiegman A, Bruckert E, Schmitt CP, et al. Efficacy and safety of lipoprotein apheresis in children with homozygous familial hypercholesterolemia: A systematic review. Vol. 13, Journal of Clinical Lipidology. Elsevier Ltd; 2019. p: 31-9.
Bibliografía recomendada
– Mata P, Alonso R, Ruiz A, González-Juanatey JR, Badimón L, Díaz-Díaz JL, et al. Diagnóstico y tratamiento de la hipercolesterolemia familiar en España: documento de consenso. Semergen. 2015; 41: 24-33.
Documento de Consenso de la Sociedad Española de Arterioesclerosis sobre Hipercolesterolemia. Es una excelente guía para conocer de manera global las bases fisiopatológicas de la enfermedad, su detección precoz, así como el diagnóstico y tratamiento.
– Wiegman A, Gidding SS, Watts GF, Chapman MJ, Ginsberg HN, Cuchel M, et al. Familial hypercholesterolæmia in children and adolescents: Gaining decades of life by optimizing detection and treatment. Vol. 36, European Heart Journal. Oxford University Press; 2015. p. 2425-37.
Documento de la Sociedad Europea de Cardiología. Realiza de forma sistemática una revisión resumida de la hipercolesterolemia familiar. A destacar los puntos claves relacionados con el cribado y el tratamiento. Es un artículo muy interesante para que el pediatra pueda tener un conocimiento global de la enfermedad.
– Mach F, Baigent C, Catapano AL, Koskina KC, Casula M, Badimon L, et al. 2019 ESC/EAS guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Atherosclerosis. 2019; 290: 140-205.
Guía de la Sociedad Europea de Cardiología sobre el enfoque de las dislipemias. Tiene especial interés el apartado de las modificaciones en el estilo de vida y el específico para la edad pediátrica.
– Plana N, Rodríguez-Borjabad C, Ibarretxe D, Masana L. Hipercolesterolemia familiar en la infancia y la adolescencia: una realidad oculta. Clin Investig Arterioscler. 2107; 29: 129-40.
Revisión sobre la hipercolesterolemia familiar en la infancia. Es un artículo muy bien estructurado, que permite al lector adquirir, de manera rápida, los conocimientos actuales sobre el diagnóstico y tratamiento.
– Ascaso JF, Mata P, Arbona C, Civeira F, Valdivielso P, Masana L. Hipercolesterolemia familiar homocigota: Adaptación a España del documento de posición del grupo de consenso sobre hipercolesterolemia familiar de la Sociedad Europea de Arteriosclerosis. Documento de Consenso de la Sociedad Española de Arteriosclerosis (SEA) y la Fundación Hipercolesterolemia Famil. Clin Investig Arterioscler. 2015; 27: 80-96.
Documento Consenso de la Sociedad Española de Arterioesclerosis sobre la hipercolesterolemia familiar homocigota, enfermedad rara y potencialmente mortal. El artículo recomienda el tratamiento con una combinación de cambios del estilo de vida, terapia hipolipemiante con fármacos y aféresis de LDL.
| Caso clínico |
|
Niña de 12 años que acude a la revisión del niño sano. Antecedentes personales: sin enfermedades previas y vacunas según calendario. Antecedentes familiares: madre de 35 años, sana, G1A0V1. Padre de 43 años, hipercolesterolemia en tratamiento con estatinas desde hace 16 años (atorvastatina 40 mg/día). Abuelos maternos sanos. Abuela paterna hipertensión arterial. Abuelo paterno fallecido de infarto agudo de miocardio a los 53 años y antecedente de hipercolesterolemia. Tío paterno con hipercolesterolemia en tratamiento, con estatinas desde hace 20 años. Examen físico: peso: 47 kg (p79, 0,84 DE). Talla: 150 cm (p59, 0,24 DE). IMC: 20,89% (p67, 0,45 DE). Superficie corporal: 1,4 m2. Tensión arterial: sistólica: 100 mmHg (p28 -0,6 DE); diastólica: 54 mmHg (p21, -0,81 DE). Sin xantomas, resto de exploración normal, Tanner III. Dados los antecedentes familiares en rama paterna, se realiza analítica donde se objetiva: colesterol total: 296 mg/dL (100-200); C-HDL: 54,3 mg/dL (35-65); C-LDL: 234,3 mg/dL (60-130); C-VLDL: 7,4 mg/dL (6,5-27); triglicéridos: 37 mg/dL (35-135). Hormonas tiroideas: TSH: 4,5 μU/l (0,64-6,2); T4L: 1,5 ng/dl (0,89-1,76). Es remitida por su pediatra a consulta de Endocrinología donde, evolutivamente se solicita el estudio genético donde se demuestra una variante en un alelo del gen del receptor de LDL, RLDL, (c.1879G>A). Se inicia tratamiento con pravastatina 10 mg al día antes de acostarse, junto con medidas higiénico-dietéticas (dieta y ejercicio físico regular).
|