 |
| Temas de FC |
A. Llort Sales, L. Gros Subias
Servicio de Oncología y Hematología Pediátrica.
Hospital Universitario Materno-Infantil Vall d’Hebron. Barcelona
| Resumen
Los tumores renales en la infancia y la adolescencia representan el 7% de las neoplasias. El tumor más frecuente es el tumor de Wilms que, en la mayoría de pacientes, se diagnostica antes de los 5 años de edad. Un 10% de los casos con tumor de Wilms se asocian a síndromes que predisponen a la enfermedad. El pronóstico, con un tratamiento multidisciplinar (cirugía, quimioterapia y radioterapia) es excelente para la mayoría de pacientes, con una supervivencia global a los 5 años del 90%. No obstante, existe un grupo de pacientes con peor respuesta al tratamiento. Cada vez se puede caracterizar mejor este grupo por aspectos clínico-histológicos; pero, sobre todo, por aspectos biológicos, que permiten ajustar de forma adecuada el tratamiento a cada paciente. Un elemento importante para estos pacientes es el seguimiento a largo plazo de los posibles efectos secundarios del tratamiento. |
| Abstract
The renal tumours affecting infants and young children account for 7% of all childhood tumours. Wilms’ tumor (WT) is the most common primary renal malignancy in children. The majority of patients with WT are diagnosed before 5 years of age. Up to 10% of cases associates to syndromes that increase risk of WT. Treatment with an multidisciplinary approach (surgery, chemotherapy and radiotherapy) achieve an excellent 5 years overall survival, 90% for all the patients. Nevertheless a group of patients have a worse response to treatment. We can characterize the patients with clinical, histological and biological aspects to adapt the treatment according to risk groups. |
Palabras clave: Tumor de Wilms; Tumores Renales; Niño; Adolescente
Key words: Wilms Tumor; Renal Tumours; Children; Adolescent
Pediatr Integral 2016; XX (7): 447-457
Tumores renales en la infancia y adolescencia
Introducción
Los tumores renales representan aproximadamente un 7% de todas las neoplasias en niños y adolescentes. El tumor de Wilms o nefroblastoma es la neoplasia renal más frecuente (75% de los tumores renales) en Pediatría, con una prevalencia de un caso por cada 10.000 niños menores de 15 años(1); aunque, en el caso de adolescentes entre 15 y 19 años, el carcinoma renal es el más frecuente(2).
Tumor de Wilms
El tumor de Wilms (TW) es el tumor renal más frecuente en la infancia, suele aparecer antes de los 5 años y es raro en mayores de 10 años.
Es el tumor renal más frecuente, que aparece a partir de focos de células metanéfricas o también llamados restos nefrogénicos. El TW es un cáncer embrionario que se gesta durante el desarrollo del tejido renal. Está formado por: células blastematosas indiferenciadas, células epiteliales inmaduras y estroma; estructuras todas ellas presentes durante la nefrogénesis normal. Así, aproximadamente, el 40% de los TW esporádicos y el 100% de los TW bilaterales pueden estar asociados a restos nefrogénicos, que se consideran lesiones precursoras de TW(3).
La supervivencia del TW ha mejorado significativamente en las pasadas décadas, pasando la supervivencia a los 5 años del 20%, en los años 1960, a un 90% en la actualidad(4).
Dicha mejora se debe fundamentalmente al abordaje multidisciplinar del tratamiento, dentro de protocolos cooperativos con el fin de optimizar el tratamiento y establecer una estratificación según el riesgo. Los principales grupos que lideran dicha mejora son, en Europa, la SIOP (Société International de Oncologie Pediatrique) y el grupo norteamericano de Oncología Pediátrica (COG) a través del NWTS (The National Wilms Tumor Study).
Estos dos grupos tienen dos aproximaciones de tratamiento distintas. El protocolo SIOP se basa en una quimioterapia preoperatoria, con el fin de reducir el riesgo de ruptura tumoral intraoperatoria y basa el riesgo en función de la respuesta a la quimioterapia recibida y la histología presente en el momento de la nefrectomía. Esto permite establecer 3 grupos de riesgo (bajo, intermedio o alto) y determinar el tratamiento postoperatorio más adecuado. Por su parte, el grupo del NWST-COG recomienda la nefrectomía de entrada, y la estratificación del tratamiento está basado en factores pronósticos, que incluyen la histología (favorable o desfavorable)(4).
Síndromes asociados con el tumor de Wilms
Varios síndromes se asocian a un mayor riesgo de TW y se debe realizar un screening específico para su detección precoz en los pacientes con estas alteraciones.
El TW aparece habitualmente en niños sanos, pero hay un 10% de casos que aparecen en niños con alguna alteración genética.
En un estudio realizado en el Institut Curie, donde se analizaron 295 casos de TW, un 17,6% presentaban alguna alteración genética. Los niños pueden tener un fenotipo identificativo (sobrecrecimiento, aniridia, malformaciones génito-urinarias). Estos síndromes se han agrupado en síndromes de hipercrecimiento y no hipercrecimiento. La tabla I muestra los distintos síndromes asociados a riesgo de TW(5).

Genómica del TW
El TW es un cáncer que se produce por un desarrollo renal aberrante. Se produce por mutaciones en varios genes. Un 33% de TW presentan mutaciones en WT1, CTNNB1 o WTX. Otro subconjunto es el resultado de mutaciones en los genes de procesamiento de miARN (micro ARN), incluyendo DICER1. Otros genes mutados de forma frecuente son: SIX1 y SIX2 (factores de transcripción claves en el desarrollo renal inicial).
Hay un elevado número de casos asociado a alteraciones genéticas, como el síndrome Denish Drash, síndrome WARG, síndrome Beckwith-Wiedemann.
Síndromes asociados a la mutación WT1
Es un gen localizado en el cromosoma 11 (11p13). Es un factor de transcripción necesario para el desarrollo genitourinario normal. Está presente en un 10-20% de los casos de TW esporádicos. La mutación WT1 germinal es más frecuente asociada a síndromes genéticos (WARG, TW bilaterales, etc.).
• El síndrome de WARG se caracteriza por: tumor de Wilms, aniridia, anomalías genitourinarias y retraso mental. Se asocia con la deleción en el cromosoma 11 (del 11p13) y representa un 0,4% de los niños con TW. Aproximadamente, un 15% de los niños con WARG tienen TW bilaterales(5,6).
• El síndrome de Denis-Drash se asocia a anomalías del desarrollo genito-urinario y esclerosis mesangial difusa. Se asocia a mutaciones “sin sentido” en la línea germinal del gen WT1. Los niños afectos de síndrome de Denish-Drash tienen un altísimo riesgo de desarrollar un TW (90%).
Síndromes asociados a la mutación WT2
Otro locus asociado al TW es el de la región 11p15.5. Cuando se presenta en la línea germinal, causa el síndrome de Beckwith-Wiedemann.
• Síndrome de Wiedemann-Beckwith es un síndrome de hipercrecimiento que se manifiesta con un crecimiento asimétrico de una o más partes del cuerpo, macroglosia, onfalocele y anomalías renales. Tiene más riesgo de presentar TW (20%) y otros tumores (rabdomiosarcoma, hepatoblastoma). Representa el 1% de los casos de TW.
• Otros síndromes asociados a TW: síndrome de Sotos, síndrome de Li Fraumeni y síndrome de Bloom.
Causas no sindrómicas de TW
Tumor de Wilms familiar: los hermanos de niños con TW tienen <1% de probabilidades de desarrollar un TW y el riesgo de TW de los descendientes de TW unilaterales (esporádicos) es <2%. Se han descrito 2 genes en el TW familiar: FTW1 y FTW2 (19q13.4). Entre los familiares, es frecuente encontrar malformaciones genitourinarias.
Hemihipertrofia: crecimiento excesivo asimétrico de una o más partes del cuerpo. Representa un 2,5% de los casos de TW. La incidencia global de TW es de 5,9%, aproximadamente.
A pesar del buen pronóstico del TW, hay un subgrupo considerado de alto riesgo, que representa aproximadamente el 25% de los casos. Este grupo incluye a los TW con histología desfavorable (anaplásicos), bilaterales o recidivas.
Algunos datos biológicos, como la pérdida de heterocigosidad (LOH) en 1p/16q se han asociado a un mayor riesgo de recidivas. No obstante, un estudio del grupo alemán de SIOP concluyó que estas alteraciones se asociaban a histología desfavorable y que, por lo tanto, la presencia de alteraciones biológicas (16q, 11q y 22q) no tenían un impacto pronóstico independiente. Por esta razón los protocolos SIOP no utilizan todavía estos biomarcadores para estratificar el riesgo. Conceden mayor peso a la respuesta histológica a la quimioterapia preoperatoria, el predominio de componente blastematoso identifica tumores más resistentes a la quimioterapia(6,7).
También son de peor pronóstico los tumores del subtipo anaplásico. Entre ambos subtipos se incluyen el 10-15 % de TW con mal pronóstico.
Clínica
En la mayoría de pacientes, el TW se presenta como una masa asintomática, se suele detectar de forma casual por los padres o el pediatra.
El TW es, en la mayoría de casos, asintomático y se detecta de forma casual por los padres, durante el baño o al vestir al niño, o en una consulta rutinaria del pediatra. Es menos frecuente que se presente con algún síntoma inespecífico, como: dolor abdominal (40%), anorexia, náuseas o vómitos, irritabilidad o malestar general. También, pueden presentar: fiebre, hematuria microscópica (24%), más raramente macroscópica (18%), e hipertensión arterial (25%) por aumento de renina(8). En ocasiones, se puede presentar con varicocele, especialmente izquierdo, por obstrucción de la vena espermática(9).
Es muy característica la exploración del abdomen de estos pacientes. Se palpa y, en ocasiones, se observa una masa en flanco, dura, pero no pétrea, habitualmente de bordes bien delimitados y que no suele atravesar la línea media (estos hallazgos suelen ser diferentes en el neuroblastoma)(10). Uno de los riesgos importantes en el TW al diagnóstico es la rotura tumoral, que puede provocar una hemorragia aguda y requerir un tratamiento quirúrgico urgente. Por este motivo, deben evitarse los traumatismos abdominales, la exploración debe realizarse de forma cuidadosa y evitar palpaciones repetidas(8). En la exploración física, debemos observar la presencia de alguna de las alteraciones asociadas a síndromes que predisponen al TW, presentes en el 10% de los pacientes(8).
Diagnóstico y estudio de extensión
Aunque el TW es el tumor renal más frecuente en la infancia, se deben descartar otras lesiones malignas (linfoma de Burkitt, leucemia, rabdomiosarcoma, otros tumores renales primarios) o procesos benignos (abscesos, hidronefrosis, enfermedad poliquística)(8,11).
En los primeros meses de vida, el tumor renal más frecuente es el nefroma mesoblástico congénito(12) y, en adolescentes, es importante descartar el carcinoma renal(2). Es importante el diagnóstico diferencial con el neuroblastoma(10). Además de por imagen, se debe realizar estudio de catecolaminas en orina, para descartar esta posibilidad.
El estudio de la enfermedad local se debe realizar con ecografía y TC o RM abdominal(11,13-15).
La ecografía es la primera exploración a realizar, no precisa sedación ni irradia. Además del riñón afecto, permite explorar: el riñón contralateral, la afectación vascular y la presencia de enfermedad extrarenal. Se presenta como una masa grande, de límites bien definidos, con una localización principalmente intrarenal. El tumor puede ser sólido y homogéneo, pero es, por lo general, heterogéneo, con áreas de hemorragia, necrosis o quistes. En el 15%, el tumor contiene calcificaciones. El tejido renal normal suele estar desplazado a la periferia del tumor(13,15).
El TC de abdomen permite valorar el tumor y su extensión loco-regional, pero si es posible, se debe sustituir por la RM para evitar la irradiación abdominal.
En RM, se aprecia un tumor heterogéneo, lobulado e hipointenso comparado con el riñón sano en secuencias T1 e hiper – o isointenso en secuencias T2. Se aprecian focos de hemorragia. Tras la administración del contraste, el tumor capta con menor avidez que el riñón adyacente. En la RM, se puede apreciar la presencia de extensión extrarenal, la presencia de adenopatías y la trombosis de la vena cava inferior. También, permite detectar focos de nefroblastomatosis en el tejido sano y en el riñón contralateral (Fig. 1)(13,14).

Figura 1. RM Nefroblastomatosis.
El problema es que requiere sedación para poder realizarla en niños pequeños (Fig. 2).

Figura 2. RM: tumor de Wilms.
La diseminación del TW puede producirse por vía hematógena y, con mayor frecuencia, a los pulmones, menos frecuente es la afectación hepática y ósea.
El estudio de las metástasis pulmonares se puede realizar con Rx de tórax y con TC sin contraste. El tratamiento como enfermedad metastásica en los pacientes que presentan nódulos en el TC, pero no visibles en la Rx, sigue siendo un dilema (Fig. 3)(16).

Figura 3. TC: metástasis pulmonares de tumor de Wilms.
El papel del FDG-PET en el estadiaje del TW no está del todo establecido(14).
En los centros americanos, el diagnóstico y el riesgo histológico se determinan en el momento inicial, con la pieza de nefrectomía. Para los protocolos europeos, el diagnóstico se puede establecer con los estudios clínico-radiológicos y no es imprescindible el estudio histológico para iniciar el tratamiento con quimioterapia preoperatoria. No obstante, algunos trabajos han demostrado la seguridad de la toma de muestras con aguja fina o Tru-cut guiadas por imagen para obtener un diagnóstico de confirmación y evitar errores. Esto es especialmente importante para pacientes en edades poco frecuentes para el TW, sobre todo adolescentes, o cuando la presentación clínica o radiológica es atípica(17,18).
El estadiaje del TW, según la SIOP, determina 5 estadios que se describen en la tabla II.

Histología
La clasificación histológica es un factor pronóstico fundamental y determina el tratamiento de los pacientes, tanto en los protocolos europeos (SIOP) como en los americanos (COG).
El TW se desarrolla a partir de restos nefrogénicos(3). El aspecto histológico es la de una lesión mixta con proporciones variables de los tres componentes celulares:
• Blastematoso, epitelial y estroma. El componente blastematoso es el elemento menos diferenciado y está formado por células pequeñas o intermedias, densamente empaquetadas, con citoplasma escaso y núcleos pequeños. Pueden mostrar distintos patrones de agrupación. El componente estromal está compuesto por células indiferenciadas mesenquimales, pero también puede mostrar: diferenciación variable a músculo liso y esquelético, células adiposas, cartílago y hueso. El componente epitelial simula estructuras del riñón embrionario, conformando rudimentos de rosetas, túbulos y glomérulos(19).
La presencia de anaplasia en el tejido tumoral es un factor de mal pronóstico e implica una peor respuesta al tratamiento. Este fenómeno se aprecia en el 5% de TW, sea de forma focal o difusa. En los protocolos americanos, la presencia de anaplasia focal o difusa se considera como factor de mal pronóstico. Para los protocolos SIOP, la presencia de anaplasia focal es considerada de riesgo intermedio, la anaplasia difusa supone alto riesgo. En la tabla III, se enumeran los grupos de riesgo histológico para los protocolos SIOP(20).

Tratamiento
El tratamiento del tumor de Wilms se basa en una aproximación multidisciplinar y estratificada según el riesgo; la quimioterapia y la cirugía son los elementos fundamentales; la radioterapia juega un papel complementario en pacientes seleccionados.
El tratamiento del TW se basa en la cirugía y en la quimioterapia. Los resultados son excelentes en una parte importante de pacientes, con supervivencias del 90%. Solo un grupo de pacientes, cada vez mejor identificados, tiene un pronóstico más desfavorable, a pesar del tratamiento más intensivo(4,8,21,22).
El progreso en la curación de estos pacientes se basa en el tratamiento dentro de estudios multicéntricos. Los resultados en supervivencia son similares en los estudios SIOP y COG. La mayor diferencia en el plan de tratamiento, está en relación al momento óptimo de la cirugía para cada uno de los grupos. Los centros americanos realizan, en general, la nefrectomía como primera parte del tratamiento, para los europeos la administración de quimioterapia preoperatoria supone beneficios importantes(4).
Cirugía
El tratamiento quirúrgico standard del TW unilateral es la ureteronefrectomía radical. Algunos aspectos quirúrgicos importantes son: la manipulación cuidadosa para evitar roturas renales intraoperatorias; intentar ligar los vasos renales antes de la manipulación del tumor para evitar diseminación hematógena; y ligar el uréter lo más distal posible. De gran importancia, es explorar todo el abdomen, incluyendo: el riñón contralateral, realizar un muestreo ganglionar y tomar biopsia de cualquier zona sospechosa.
En algunos casos, el tumor se extiende, en forma de trombo tumoral, a la vena renal, la vena cava y, más raramente, alcanza la aurícula(23). El manejo quirúrgico, cuando persiste un trombo por encima de la cava hepática, es muy complejo y con alto riesgo de complicaciones quirúrgicas.
La cirugía puede jugar un papel en el tratamiento de las metástasis, sobre todo pulmonares y hepáticas, especialmente en lesiones que persisten tras 6 semanas de tratamiento preoperatorio(23).
En los últimos años, se ha planteado la posibilidad de cirugía conservadora (NSS, nephron sparing surgery)(24) y cirugía por vía laparoscópica(23,25). En las dos situaciones, se deben valorar de forma cuidadosa las ventajas y riesgos de la técnica, se deben cumplir criterios muy estrictos de selección de pacientes y, sobre todo, se deben realizar en centros de referencia con experiencia para realizar estos procedimientos.
Radioterapia
El TW es un tumor muy radiosensible; no obstante, en muchos pacientes, la combinación de quimioterapia y cirugía es suficiente para garantizar la curación.
En un grupo seleccionado de pacientes, la radioterapia sigue teniendo un papel fundamental para mejorar los resultados del tratamiento y aumentar la supervivencia.
Las indicaciones actuales de irradiación son:
• Irradiación del flanco afecto en pacientes en estadio III y de riesgo histológico intermedio. En pacientes con ganglios positivos y/o enfermedad residual microscópica, se administran 10,8 Gy. Si persiste enfermedad residual macroscópica, se realiza un boost sobre el resto hasta 25,2 Gy. En pacientes con estadio III por rotura tumoral, se deben administrar 15 Gy en todo el abdomen, si hay implantes peritoneales macroscópicos, la dosis sobre el abdomen es de 20 Gy y boost hasta 25,2 Gy en los restos macroscópicos.
• Los tumores de alto riesgo histológico (anaplásico) estadios II y III reciben 10,8 Gy.
• La radioterapia pulmonar se administrará a los pacientes con metástasis pulmonares que no han respondido al tratamiento con quimioterapia preoperatoria. Se administran 12 Gy en ambos campos pulmonares y hasta 25-30 Gy en lesiones residuales macroscópicas (Fig. 4)(26).
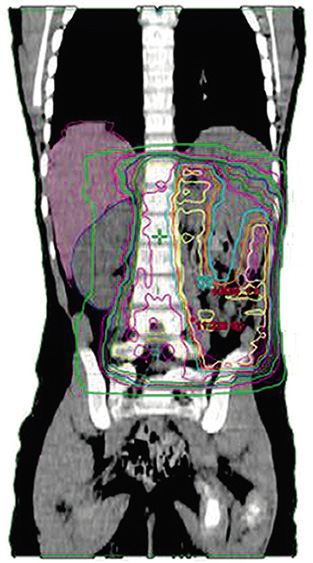
Figura 4. Dosimetria RTX.
Quimioterapia
El TW es un tumor muy quimiosensible. La base del tratamiento farmacológico es la combinación de vincristina, actinomicina-D para pacientes de bajo riesgo y riesgo intermedio y de ciclofosfamida, adriamicina, etopósido y carboplatino para los pacientes de alto riesgo.
En los protocolos europeos (SIOP), el tratamiento prequirúrgico se establece en dos niveles. Los pacientes con enfermedad localizada reciben 4 semanas de tratamiento con vincristina y actinomicina-D. Los pacientes con metástasis al diagnóstico reciben quimioterapia durante 6 semanas con la combinación: vincristina, actinomicina-D y adriamicina. El objetivo de esta quimioterapia preoperatoria es reducir las complicaciones operatorias y evaluar la sensibilidad del tumor a la quimioterapia.
El plan de quimioterapia adyuvante está determinado en los protocolos europeos por el riesgo histológico de la pieza de nefrectomía y por el estadiaje postquirúrgico.
En la tabla IV, se resumen los fármacos y la duración del tratamiento para cada grupo de riesgo en el protocolo SIOP Nephroblastoma-01.

Un grupo de pacientes con enfermedad localizada e histología de bajo riesgo no reciben tratamiento citostático tras la cirugía. Los pacientes con estadio I y riesgo intermedio reciben 4 semanas de tratamiento con vincristina y actinomicina-D. Para pacientes con bajo riesgo histológico y estadios II y III, el tratamiento se basa en vincristina y actinomicina-D durante 27 semanas. Uno de los objetivos iniciales de este estudio era discernir si el uso de adriamicina en los pacientes de riesgo histológico intermedio en estadios II y III aportaba mejoría en la supervivencia. Por este motivo, se randomizó el tratamiento de este grupo entre 2 fármacos (vincristina + actinomicina-D durante 27 semanas) o 3 fármacos (vincristina + actinomicina-D + adriamicina durante 27 semanas). Tras analizar 583 pacientes con un seguimiento de más de 60 meses, se obtuvo una supervivencia global a 5 años de 96,5% para pacientes que recibieron adriamicina y de 95,8% para los que no la recibieron. Con estos resultados, se cerró la randomización y se estableció el tratamiento de estos pacientes con 2 fármacos(27). Los pacientes en estadio III reciben la radioterapia correspondiente.
Los pacientes de alto riesgo histológico son los pacientes con peor pronóstico, por lo tanto, deben recibir tratamiento más intensivo. Los pacientes en estadio I reciben 3 fármacos durante 27 semanas. Los pacientes en estadios II y III reciben un régimen de tratamiento con 4 fármacos, con ciclos alternos de ciclofosfamida y adriamicina con otros de carboplatino y etopósido hasta completar 34 semanas de tratamiento.
Los pacientes con enfermedad metastásica al diagnóstico reciben quimioterapia postoperatoria, según 2 criterios: el riesgo histológico y el estadiaje local del tumor primario, y la respuesta de las metástasis al tratamiento neoadyuvante. Los pacientes con remisión completa de las metástasis, por respuesta a quimioterapia preoperatoria o por cirugía, reciben tratamiento según el estadio local (3 fármacos AVD, excepto los de histología de alto riesgo que reciben 4 fármacos). Los pacientes en estadio III reciben radioterapia en flanco o abdomen y los que precisan cirugía de las metástasis, irradiación pulmonar. Los pacientes con persistencia de las metástasis reciben 4 fármacos con radioterapia pulmonar en todos los casos, y local según estadiaje del primario.
Tratamiento de TW bilateral. Estadio V
Entre el 4 y el 13% de niños afectos de TW presentan afectación bilateral. Estos pacientes, con frecuencia, presentan: restos nefrogénicos, malformaciones o síndromes de predisposición. El gran reto para los equipos multidisciplinares consiste en obtener la curación de estos pacientes manteniendo la mayor capacidad funcional renal posible, que permita un desarrollo y crecimiento normal. Son de gran importancia los estudios radiológicos para monitorizar la respuesta y para el seguimiento. El planteamiento terapéutico consiste en administrar quimioterapia preoperatoria hasta obtener la máxima respuesta. Cuando se considere el momento óptimo, se debe priorizar la cirugía conservadora (nephron sparing surgery). Se debe consolidar con quimioterapias ajustadas al riesgo y estadiaje más elevado(28).
Tratamiento de las recaídas
El porcentaje de recaídas en TW es bajo y, sobre todo, suelen afectar a pacientes con histología desfavorable, aunque un 15% afecta a pacientes con histología favorable.
Debido al número bajo de casos, en esta situación, es difícil establecer tratamientos estandarizados. El tratamiento se debe basar en fármacos que no se utilizaran en los esquemas de primera línea. Incluso se ha utilizado la quimioterapia a altas dosis con rescate con progenitores hematopoyéticos(29).
Los pacientes que recaen tras recibir 2 fármacos, pueden rescatarse con regímenes de 4 fármacos y tratamiento local, con supervivencias del 70-80%. Por el contrario, los pacientes con histología desfavorable (anaplasia difusa y tipo blastematosos post-quimioterapia) que recibieron 4 drogas son pacientes con mal pronóstico. Se pueden tratar con nuevas combinaciones de fármacos o incluirlos en estudios fase I-II. La supervivencia se sitúa alrededor del 10%.
Tumores renales no Wilms
Aunque el TW es el tumor renal más frecuente en la infancia, es importante considerar el diagnóstico de otro tipo de tumores renales menos frecuentes que requieren un plan de tratamiento distinto.
La edad de diagnóstico, la forma de presentación clínica, el comportamiento biológico y la imagen radiológica pueden ser diferentes para cada tipo de tumor. En la tabla V, se resumen las características clínicas y radiológicas de los tumores pediátricos más frecuentes en la primera década de la vida(14).

Sarcoma de células claras
Representa entre el 3 y el 5% de todos los tumores renales. Es más frecuente en niños entre 2 y 4 años. Es un tumor agresivo y puede metastatizar a hueso y cerebro. La presentación clínica suele ser: dolor abdominal, hematuria e hipertensión arterial. Radiológicamente, puede confundirse con otros tumores renales (TW, nefroma mesoblástico congénito o tumor rabdoide).
Se han descrito alteraciones citogenéticas, como: la traslocación t(10;17)(q22;p13/p12)53 y la delección de 14q24q31.
El tratamiento se basa en la cirugía con nefrectomía y quimioterapia intensiva que incluya doxorubicina.(2,30)
Carcinoma renal (CR)
Representa el 2-6% de los tumores renales pediátricos, aunque es el tumor renal más frecuente en adolescentes. La edad de presentación suele ser 10-11 años. El CR se origina de las células epiteliales del túbulo renal. Existen diversos subtipos histológicos de CR.
La presentación clínica suele ser: dolor abdominal, hematuria y masa palpable abdominal. A diferencia del TW, es poco frecuente que se manifieste como una masa asintomática.
Es más frecuente el CR en supervivientes de cáncer infantil y en determinados síndromes, como: enfermedad Von-Hippel-Lindau, esclerosis tuberosa, en patología quística renal, en fases terminales de enfermedad renal, en enfermedad drepanocítica y en trasplantados renales pediátricos.
Puede metastatizar a diferentes sitios, preferentemente a pulmón (40%), huesos o hígado.
El diagnóstico tiene que ser siempre histológico. Radiológicamente, suele presentarse como una masa más pequeña que el TW y es frecuente la invasión vascular y la afectación linfadenopática (Fig. 5).
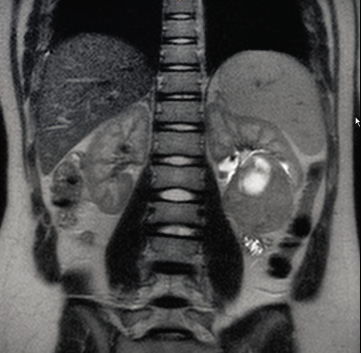
Figura 5. Carcinoma renal.
Puede demostrarse frecuentemente, una pérdida de heterocigosidad 22q11 y q12 y pérdida de la expresión de la proteína SMARCB1.
El factor pronóstico más importante es el estadio al diagnóstico, siendo de alrededor de 90% en el estadio I, del 50-80% en los estadios II y III y menos del 10% en el estadio IV.
La base del tratamiento es la nefrectomía radical junto con linfadenectomía regional. El CR es un tumor poco sensible a la quimioterapia y radioterapia. Se han utilizado tratamientos inmunológicos, como Interferón-a y interleukina-2 y, más recientemente, diversas terapias dirigidas, como: sorafenib, sunitinib, bevacizumab, temsirolimus, everolimus y pazopamid; aunque, en pediatría hay pocos datos testados(2,30).
Nefroma mesoblástico
Es el tumor renal más frecuente en los 3 primeros meses de vida. El 90% se diagnostica antes del año de vida. En la actualidad, el diagnóstico es prenatal en muchos casos. Se presenta como una masa abdominal, a veces, acompañada de hematuria. Puede presentarse con hipertensión arterial y con hipercalcemia. Se distinguen dos variantes histológicas: la clásica y la celular. En esta última, se ha identificado una traslocación, t(12;15)(p13;q25), que genera una proteína de fusión ETV6-NTRK3 implicada en las vías Ras-RAF y PI3K-Akt.
En la mayoría de casos, el tratamiento es quirúrgico, con nefrectomía, pero un pequeño porcentaje puede presentar recaídas locales, e incluso metástasis, que requieren tratamiento con quimioterapia(2,30).
Tumor teratoide rabdoide atípico
Hasta 1981, se consideró como una variante de TW. Supone el 2% de los tumores renales. Afecta a niños pequeños, el 80% se diagnostica antes de los 2 años. Se presenta como masa abdominal, que suele acompañarse de hematuria y, a veces, de hipercalcemia. Con frecuencia, los primeros síntomas se producen por la presencia de metástasis en pulmones, hígado o cerebro. Se asocia a mutaciones germinales o delecciones del gen SMARCB1/INI1/SNF5/BAF47 en el cromosoma 22q11.2.
Es un tumor muy agresivo con mal pronóstico. La combinación de distintos fármacos, con cirugía y radioterapia, incluso el uso de quimioterapia a altas dosis, ha permitido discretos avances en la supervivencia(2,30).
Seguimiento a largo plazo
Los supervivientes de tumores renales deben seguir un seguimiento específico a muy largo plazo por el riesgo de desarrollar insuficiencia renal. Los estudios experimentales sugieren que las nefronas restantes, sufren un estado crónico de hiperfiltración, que pueden evolucionar a: uremia, proteinuria y glomeruloesclerosis. Los pacientes con menos de un 50% de nefronas suelen tener la función renal normal, pero tienen riesgo de desarrollar hipertensión arterial o microalbuminuria. Por este motivo, debemos realizar una monitorización de: microalbuminuria, proteinuria, tensión arterial y función renal (creatinina y filtrado glomerular). Hay que tener presente en el seguimiento: el tratamiento recibido, especialmente si ha habido radioterapia, vigilancia de la zona irradiada, o han recibido antraciclínicos (monitorización de la función cardíaca).
El plan de seguimiento tiene 2 fases diferenciadas:
1. Fase 1: orientada a detectar recidivas precoces.
2. Fase 2: orientada a detectar efectos secundarios:
• Al final del tratamiento: exploración física, ecografía abdominal, Rx/Tac tórax, orina, función (fx) renal, audiometría y ecocardiograma.
• Primer año post-tratamiento (controles cada 8 semanas), exploración física, ecografía abdominal, Rx tórax, orina y fx renal.
• Segundo y tercer año post-tratamiento (controles cada 4 meses): exploración física, ecografía abdominal, Rx tórax, orina y fx renal.
• Cuarto año (controles cada 6 meses): exploración física, ecografía abdominal, Rx/Tac tórax, orina, fx renal, audiometría y ecocardiograma.
• Años 5-10: controles cada 6 meses: exploración física y orina.
Bibliografía
Los asteriscos reflejan el interés del artículo a juicio del autor.
1.*** Breslow N, Olshan A, Beckwith JB, et al. Epidemiology of Wilms tumor. Med Pediatr Oncol. 1993; 21: 172-81.
2.*** Ahmed HU, Arya M, Levitt G, et al. Part I: Primary malignant non-Wilms’ renal tumours in children. Lancet Oncol. 2007; 8: 730-7.
3.** Beckwith JB, Kiviat NB, Bonadio JF. Nephrogenic rests, nephroblastomatosis, and the pathogenesis of Wilms’ tumor. Pediatr Pathol. 1990; 10: 1-36.
4.*** Dome JS, Graf N, Geller JI, et al. Advances in wilms tumor treatment and biology: Progress through international collaboration. J Clin Oncol. 2015; 33: 2999-3007.
5.** Dumoucel S, Gauthier-Villars M, Stoppa-Lyonnet D, et al. Malformations, genetic abnormalities, and Wilms tumor. Pediatr Blood Cancer. 2014; 61: 140-4.
6.** Royer-Pokora B, Beier M, Henzler M, et al. Twenty-four new cases of WT1 germline mutations and review of the literature: genotype/phenotype correlations for Wilms tumor development. Am J Med Genet A. 2004; 127A: 249-57.
7.*** Klamt B, Schulze M, Thäte C, et al. Allele loss in Wilms tumors of chromosome arms 11q, 16q, and 22q correlate with clinicopathological parameters. Genes Chromosomes Cancer. 1998; 22: 287-94.
8.** Fernandez C, Geller JI, Ehrlich PF, et al. Renal Tumors. In: Pizzo PA, Poplack DG, Adamson PC, et al, editors. Principles and Practice of Pediatric Oncology. 7th ed. Philadelphia: Wolters-Kluwer; 2016. p. 753-71.
9.** Vizmanos D, Gallego S, Gros L, et al. Lactante de 9 meses con aumento unilateral del tamaño testicular. An Pediatría. 2005; 63: 569-70.
10.*** Dumba M, Jawad N, McHugh K. Neuroblastoma and nephroblastoma: A radiological review. Cancer Imaging. Cancer Imaging; 2015; 15: 1-14.
11.*** Malkan AD, Loh A, Bahrami A, et al. An Approach to Renal Masses in Pediatrics. Pediatrics. 2015; 135: 142-58.
12.*** Lowe LH, Isuani BH, Heller RM, et al. Pediatric renal masses: Wilms tumor and beyond. Radiographics. 2000; 20: 1585-603.
13.*** Smets AM, de Kraker J. Malignant tumours of the kidney: imaging strategy. Pediatr Radiol. 2010; 40: 1010-8.
14.*** Decade PT, Chung EM, Conran RM. Renal Tumors of Childhood: Radiologic-Pathologic Correlation. Radiographics. 2016; 36: 499-522.
15.** Fufezan O, Asavoaie C, Blag C, et al. The role of ultrasonography for diagnosis the renal masses in children. Pictorial essay. Med Ultrason. 2011; 13: 59-71.
16.*** Smets AMJB, Tinteren H Van, Bergeron C, et al. The contribution of chest CT-scan at diagnosis in children with unilateral Wilms’ tumour. Results of the SIOP 2001 study. Eur J Cancer. Elsevier Ltd; 2012; 48: 1060-5.
17.*** Vujani? GM, Kelsey A, Mitchell C, et al. The role of biopsy in the diagnosis of renal tumors of childhood: Results of the UKCCSG Wilms Tumor Study 3. Med Pediatr Oncol. 2003; 40: 18-22.
18.** Cost NG, Granberg CF, Schlomer BJ, et al. Single institution experience with Tru-Cut renal mass biopsy for diagnosing Wilms tumor. Urol J. 2013; 10: 780-3.
19.*** Al-hussain T, Ali A. Wilms Tumor: An Update. Adv Anat Pathol. 2014; 21: 166-73.
20.*** Szychot E, Apps J, Pritchard-jones K. Wilms ’ tumor: biology , diagnosis and treatment. Transl Pediatr. 2014; 3: 12-24.
21.*** Geller JI. Current standards of care and future directions for “high-risk” pediatric renal tumors: Anaplastic Wilms tumor and Rhabdoid tumor. Urol Oncol Semin Orig Investig. Elsevier; 2015; 34: 1-7.
22.*** Dome JS, Perlman EJ, Graf N. Risk stratification for wilms tumor: current approach and future directions. Am Soc Clin Oncol Educ Book. 2014; 215-23.
23.*** Kieran K, Ehrlich PF. Current surgical standards of care in Wilms tumor. Urol Oncol Semin Orig Investig. Elsevier; 2015; 34: 1-11.
24.** Vanden Berg RNW, Bierman EN, Noord M Van, et al. Nephron-sparing surgery for Wilms tumor: A systematic review. Urol Oncol. Elsevier; 2015; 34: 24-32.
25.*** Malkan AD, Loh AHP, Sandoval JA. Minimally invasive surgery in the management of abdominal tumors in children. J Pediatr Surg. 2014; 49: 1171-6.
26.*** Verschuur A, Van Tinteren H, Graf N, et al. Treatment of pulmonary metastases in children with stage IV nephroblastoma with risk-based use of pulmonary radiotherapy. J Clin Oncol. 2012; 30: 3533-9.
27.*** Pritchard-Jones K, Bergeron C, De Camargo B, et al. Omission of doxorubicin from the treatment of stage II-III, intermediate-risk Wilms’ tumour (SIOP WT 2001): An open-label, non-inferiority, randomised controlled trial. Lancet. Pritchard-Jones et al.; 2015; 386: 1156-64.
28.*** Aronson DC, Slaar A, Heinen RC, et al. Long-Term Outcome of Bilateral Wilms Tumors (BWT). Pediatr Blood Cancer. 2011; 1110-3.
29.** Ha TC, Spreafico F, Graf N, et al. An international strategy to determine the role of high dose therapy in recurrent Wilms’ tumour. Eur J Cancer. 2013; 49: 194-210.
30.*** Ahmed HU, Arya M, Levitt G, et al. Part II: Treatment of primary malignant non-Wilms ’ renal tumours in children. Lancet Oncol. 2007; 8: 842-8.
Bibliografía recomendada
– Davidoff AM. Wilms’ tumor. Curr Opin Pediatr. 2009; 21: 357-64.
Revisión de los aspectos generales y, sobre todo, del tratamiento del TW, centrado en los distintos protocolos de tratamiento del NWTS-COG. Revisión de los aspectos quirúrgicos.
– Davidoff AM. Wilms Tumor. Adv Pediatr. Elsevier Inc. 2012; 59: 247-67.
Artículo de revisión sobre el TW. Revisa aspectos de la biología, el diagnóstico de forma más completa que el anterior. También, revisa el tratamiento desde los 2 grandes grupos (NWTS-COg y SIOP).
– Gleason JM, Lorenzo AJ, Bowlin PR, et al. Innovations in the management of Wilms’ tumor. Ther Adv Urol. 2014; 6: 165-76.
Revisión del tratamiento del TW según los 2 grandes grupos, valorando los beneficios de cada uno de los abordajes. Revisa: los efectos secundarios a largo plazo, las controversias actuales y los nuevos conocimientos en la biología que pueden guiar los futuros tratamientos.
– Green DM. The evolution of treatment for Wilms tumor. J Pediatr Surg. 2013; 48: 14-9.
Revisión de la evolución del tratamiento del TW. Como elemento más interesante, la revisión de la cirugía de preservación de nefronas (NSS).
– Nakamura L, Ritchey M. Current management of wilms’ tumor. Curr Urol Rep. 2010; 11: 58-65.
Revisión del tratamiento del TW desde el punto de vista de ambos grupos internacionales. Como elemento interesante, la introducción del concepto de screening en los pacientes con síndromes de predisposición a TW.
– Pietras W. Advances and Changes in the Treatment of Children with Nephroblastoma. Adv Clin Exp Med. 2012; 21: 809-20.
Revisión del tratamiento del TW, con valoración de los planteamientos de los dos grandes grupos cooperativos internacionales.
| Caso clínico |
|
Lactante de 3 meses, acude a urgencias porque la madre durante el baño le palpa una masa abdominal sin otra sintomatología acompañante. En la exploración física, se aprecia una masa en flanco izquierdo . Se solicita en ese momento, una ecografía abdominal donde se visualiza un tumor quístico retroperitoneal izquierdo de 7,7 x 7,2 x 6,9 cm, en relación tercio superior de riñón, sugestivo de tumoración de origen renal, sin poderse descartar neuroblastoma. RMN renal para completar estudio, que muestra una voluminosa masa tumoral de origen renal izquierdo, con primera posibilidad diagnóstica el nefroma mesoblástico versus tumor de Wilms. Se realiza estudio de extensión (TC torácico) que no muestra afectación pulmonar.
Se realiza, bajo anestesia general: nefrectomía izquierda, adrenalectomía izquierda, muestreo ganglionar extenso y se reseca un pequeño fragmento de diafragma íntimamente adherido. La anatomía patológica muestra nefroblastoma blastematoso estadio local II, invasión de suprarrenal, adenopatías negativas y ausencia de invasión diafragmática. Riesgo intermedio. Inicia tratamiento quimioterápico con vincristina y actinomicina D, según protocolo de TW. A los 3 meses de tratamiento quimioterápico, acude nuevamente a urgencias por dificultad respiratoria. A la exploración física, destaca hipofonesis derecha que mostró una recidiva metastásica con afectación pleuro-pulmonar. Se inició tratamiento con quimioterapia de rescate. La paciente completó el tratamiento quimioterápico de 2ª línea. En la evaluación de la respuesta, se evidencia una gran reducción de la masa mayor, pero persisten las metástasis pulmonares bilaterales. Durante estos tres meses, a la madre (23 años de edad) le han diagnosticado de un cáncer de mama. Se revisan los antecedentes familiares y el abuelo materno fue tratado de un osteosarcoma en la tibia a los 20 años. Se derivó para estudio genético familiar.
|





