 |
| Temas de FC |
D. González-Lamuño
Profesor Titular de Pediatría. Unidad de Nefrología-Metabolismo Infantil. Servicio de Pediatría. Hospital Universitario M. Valdecilla-Universidad de Cantabria. Santander
| Resumen
La hipercalciuria es una excreción urinaria de calcio superior a 4 mg/kg/día, en el contexto de una dieta normal en calcio, proteínas y sodio. La calciuria puede estimarse en una orina aislada, determinando la relación calcio/creatinina. Por encima del año, una relación >0,2 sugiere hipercalciuria. La mayoría de las hipercalciurias están asociadas a una hipercalcemia, siendo la causa más frecuente en el niño la hiperdosificación de vitamina D. Otras causas de hipercalciuria secundaria son el hiperparatiroidismo, la inmovilización prolongada, el síndrome de Cushing, tratamiento con corticosteroides, síndrome de Fanconi, síndrome de Bartter o la acidosis tubular distal. La hipercalciuria también se puede observar en la enfermedad de Dent y asociada al raquitismo hipofosfatémico. |
| Abstract
Hypercalciuria is a urinary calcium excretion more than 4 mg/kg/day, in the context of a normal diet in calcium, protein and sodium. The calciuria can be estimated in a isolated urine by determining the calcium/creatinine ratio. A ratio >0.2 suggests hypercalciuria. Most hypercalciuria are associated with hypercalcemia, the most common being the child hiperdosificación vitamin D. Other causes are secondary hypercalciuria hyperparathyroidism, prolonged immobilization, Cushing syndrome, treatment with corticosteroids, Fanconi syndrome, Bartter syndrome and distal renal tubular acidosis. Hypercalciuria can also be observed in Dent’s disease and associated with hypophosphatemic rickets. |
Palabras clave: Hipercalciuria; Tubulopatías; Nefrocalcinosis; Litiasis renal.
Key words: Hypercalciuria; Tubulopathies; Nephrocalcinosis; Renal stones.
Pediatr Integral 2013; XVII(6): 422-432
Hipercalciuria
Introducción
La hipercalciuria se define como una excreción urinaria de calcio superior a 4 mg/kg/día, en el contexto de una dieta normal en cuanto al contenido de calcio, proteínas y sodio. La calciuria también puede ser estimada en una orina aislada, determinando la relación calcio/creatinina (Ca/Cr), expresada en mg/mg. Por encima del año de edad, una relación superior a 0,2 mg/mg sugiere hipercalciuria. Durante los primeros 6 meses de vida, los valores considerados normales son inferiores a 0,8 mg/mg y, desde los 6 meses al año de vida, de 0,6 mg/mg.
La hipercalciuria se define como una excreción urinaria de calcio superior a 4 mg (o 0,1 mmol) por cada kg de peso y día, en el contexto de una dieta normal en cuanto al contenido de calcio, proteínas y sodio. La calciuria también puede ser estimada en una orina aislada, determinando la relación calcio/creatinina (Ca/Cr), expresada en mg/mg o mmol/mmol. Por encima del año de edad, una relación superior a 0,2 mg/mg sugiere hipercalciuria.
Es complejo definir los valores de calciuria con potencial significado patológico, ya que existe una gran variabilidad en la incidencia de patología asociada a esta alteración y a que los niveles de calciuria son extremadamente variables en distintas comunidades(1). Se entiende que los límites máximos de calciuria diaria normal son, de forma absoluta, de 250 mg para la mujer y de 300 mg para el hombre y, en la edad pediátrica, de 4 mg/kg/día a partir de los 2-3 años. Durante la lactancia, las cifras de calciuria consideradas normales son significativamente más elevadas, ya que la excreción de calcio urinario es mayor en lactantes y niños pequeños que durante la edad escolar. La hipercalciuria se define, por tanto, como una excreción diaria de calcio superior a 4 mg, o 0,1 mmol, por cada kg de peso, objetivada en varias ocasiones. De forma práctica, si no es posible realizar una recogida de orina de 24 horas, las cifras de calciuria pueden estimarse a partir de las concentraciones de calcio (Ca) y creatinina (Cr) obtenidas en una muestra aislada de orina, valorando la proporción o relación entre ambas. Una relación Ca/Cr, expresada en mg/mg superior a 0,2, sugiere hipercalciuria, teniendo en cuenta que los valores normales durante los primeros 6 meses de vida son hasta de 0,8 y, desde los 6 meses al año de vida, de 0,6(2). Expresados los valores en mmol/mmol, la proporción normal es menor de 2,4 mmol/mmol en el lactante y menor de 0,6 en el niño mayor(3) (Tabla I).

La excreción de calcio urinario está aumentada en lactantes e influenciada por la fuente de leche o tipo de fórmula. Los lactantes alimentados con leche materna presentan una mayor excreción de calcio urinario; mientras que, los lactantes alimentados con fórmulas de soja tienen la menor excreción de calcio urinario. La excreción de calcio no parece verse afectada por aspectos ligados al sexo o a componentes raciales, pero está afectada por factores geográficos y/o culturales. Esto se debe a que la influencia de la dieta en la calciuria es muy importante, tanto por su contenido en calcio como en otros nutrientes, como el sodio o las proteínas. La relación Ca/Cr puede aumentar hasta en un 40%, índice Ca/Cr de 0,28, tras una comida(4). Por tanto, en la evaluación de un niño con posible hipercalciuria debe recogerse de forma exhaustiva la historia dietética, con el fin de valorar si los factores dietéticos están en relación con la calciuria.
La “hipercalciuria idiopática”, es un defecto metabólico caracterizado por alteraciones en el transporte de calcio a nivel intestinal, renal y del hueso, que se identifica con frecuencia en la población pediátrica y, especialmente, en adultos con litiasis cálcica y osteoporosis. Este trastorno familiar es debido a diferentes combinaciones de factores genéticos y dietéticos.
Una excesiva eliminación urinaria de calcio en ausencia de hipercalcemia o de otras causas conocidas de hipercalciuria son las denominadas “hipercalciurias idiopáticas”, con calciurias superiores a 6 mg/kg/24 horas. Estas alteraciones metabólicas pueden normalizarse con la disminución de la ingesta de calcio; si bien, a veces necesita la asociación de un natriurético. Una vez descartadas las causas secundarias de hipercalciuria y se establece el diagnóstico de hipercalciuria primaria (Tabla II), el paso siguiente es evaluar si las modificaciones de la dieta pueden normalizar la excreción de calcio. Si las muestras de orina se han realizado en el contexto de una dieta rica en sodio, debe recogerse una nueva muestra de orina tras 2-4 semanas de restricción sódica (menos de 2-3 g de sal al día).
Dado el carácter genético de muchas condiciones asociadas a la hipercalciuria primaria, la posibilidad de incorporar estudios genéticos a la práctica clínica ha ido modificando la categorización de estas condiciones clínicas, con un menor peso de la denominación de idiopática.

Hipercalciurias con hipercalcemia
El hiperparatiroidismo primario por adenoma paratiroideo es raro en el niño y no suele observase antes de la adolescencia. Su presencia en sujetos jóvenes obligaría a investigar una “neoplasia endocrina múltiple” de tipo I.
Entre las causas genéticas de hipercalcemia del lactante que pueden causar una nefrocalcinosis, no es raro el síndrome de Williams-Beuren, un síndrome genético por microdeleción que asocia facies característica, retraso mental y cardiopatía, debido a una deleción del cromosoma 7 que incluye el gen de la elastina. La hipercalciuria formaba parte de los criterios clínicos que definieron el síndrome, aunque posteriormente se ha documentado que no es un elemento crítico asociado al síndrome. También, se han descrito casos de nefrocalcinosis con hipercalcemia e hipercalciuria en la trisomía 21.
Se describe hipercalcemia e hipercalciuria en algunos síndromes de malabsorción intestinal de azúcares y en los hipotiroidismos no tratados. Por último, en los casos en los que no se identifica una causa evidente, y que se agrupan en las denominadas “hipercalcemias infantiles idiopáticas”, a menudo complicadas con nefrocalcinosis, comienzan a identificarse alteraciones hereditarias del metabolismo de la vitamina D, que parece jugar un papel importante. Sin embargo, debe considerarse que, en la edad pediátrica, las causas más frecuentes de hipercalcemia son las iatrogénicas: sobrecarga de vitamina D, vitamina A y calcio, sobre todo en caso de nutrición parenteral total (Tabla III).

Hipercalciurias sin hipercalcemia
Enfermedades genéticas generadoras de hipercalciuria
Estudios de las formas monogénicas de nefrolitiasis hipercalciúrica, como la acidosis tubular distal, el síndrome de Bartter (alcalosis hipopotasémica, ocasionalmente asociado con sordera), la enfermedad de Dent (con proteinuria tubular), la hipomagnesemia familiar con hipercalciuria, la nefrolitiasis hipercalciúrica con hipofosfatemia y la hipercalciuria hipocalcémica autosómica dominante, han ayudado a identificar un número significativo de transportadores, canales y receptores involucrados en la regulación de la resorción tubular de calcio(5).
Otras enfermedades hereditarias que presentan una hipercalciuria con calcemia normal son: síndrome de Lowe (síndrome oculocerebrorrenal), raquitismo hipofosfatémico con hipercalciuria, síndromes de Toni-Debré-Fanconi y seudohipoaldosteronismo(6). Nos referimos a continuación a las formas más significativas:
1. La acidosis tubular distal (antes llamada “acidosis de Albright”), en sus diferentes formas genéticas, autosómicas recesivas (con sordera asociada) o autosómicas dominantes, provoca una nefrocalcinosis medular a través de la hipercalciuria debida a la acidosis crónica. Una excreción disminuida de citrato urinario debería alertar al clínico de la posibilidad de una acidosis tubular renal distal como causa de la hipercalciuria. En ausencia de tratamiento, de forma clásica ofrece una imagen de “granos de mijo”, marcando los cálices, dibujando, en ocasiones, los contornos de la papila. En los casos menos graves, la nefrocalcinosis es más discreta, con pequeños granos opacos medulares o sólo detectables en la ecografía. Un tratamiento alcalinizante precoz que normalice la calciuria evita la aparición de las formas graves de hipercalciuria (Fig. 1).
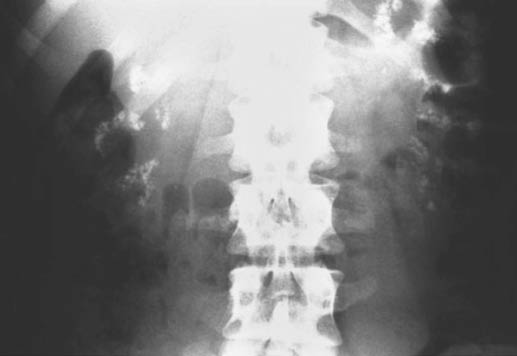
Figura 1. Ejemplo de nefrocalcinosis medular en la acidosis tubular distal.
2. El síndrome de Bartter, una enfermedad autosómica recesiva debida a mutaciones en el co-transportador de Na-K-Cl (NKCC2), el canal medular regulador de potasio (ROMK), el canal de cloro dependiente del voltaje CLC-Kb, la subunidad beta CLC-Kb o bartrina, o el receptor calcio sensible (CaSR) asociado a sordera, se acompaña de una hipercalciuria a menudo generadora de nefrocalcinosis y difícilmente controlable a pesar del uso de antiinflamatorios no esteroideos. Es la causa por la cual esta tubulopatía, caracterizada por una alcalosis hipopotasémica es, en la actualidad, una de las causas más frecuentes de nefrocalcinosis en el niño.
3. La enfermedad de Dent o “nefrolitiasis ligada al X”, donde la hipercalciuria está asociada a una proteinuria tubular de bajo peso molecular, es mucho más rara en el niño. Está asociada a mutaciones del gen que codifica un canal cloro, CLCN5 o, más raramente, del gen OCRL1 igualmente situado en el cromosoma X; OCRL1 es, con más frecuencia, responsable del síndrome de Lowe (oculocerebrorrenal), donde la hipercalciuria raramente destaca entre las pérdidas tubulares.
4. La hipomagnesemia familiar con hipercalciuria y nefrocalcinosis, enfermedad autosómica recesiva asociada a mutaciones de genes que codifican las “paracelinas” (Claudin 16 y 19)(7). Los miembros de la familia de proteínas de membrana claudinas, forman las uniones que conforman las barreras intercelulares de una gran variedad de epitelios. Estudios de estos genes han permitido conocer muchos aspectos de los mecanismos que regulan la resorción de calcio en el túbulo renal y predisponen a la hipercalciuria y nefrolitiasis.
5. El raquitismo hipofosfatémico con hipercalciuria, asociado a mutaciones autosómicas recesivas del gen SLC34A3, que codifica el co-transportador sodio-fosfato NaPi2c, puede complicarse con una nefrocalcinosis, así como la deleción del gen SLC34A1 en el marco de un síndrome de genes contiguos.
6. La hipercalciuria con hipocalcemia es excepcional. Es el resultado de un hipoparatiroidismo con defecto de reabsorción tubular del calcio por mutación activadora del receptor sensible al calcio. Se suele observar fácilmente en el periodo neonatal, con una hipercalciuria y una nefrocalcinosis asociada a grados variables de hipocalcemia.
7. Se han descrito casos de nefrocalcinosis por hipercalciuria en otras enfermedades hereditarias, entre ellas: cistinosis, enfermedad de Wilson y glucogenosis de tipo 1.
Otras hipercalciurias normocalcémicas
Son el resultado de una reabsorción ósea y/o hiperabsorción intestinal. Se trata de situaciones poco comunes: hipervitaminosis D, aporte alimentario excesivo de calcio (productos lácteos), aportes inapropiados de calcio o de fosfato en nutrición parenteral, corticoterapia, inmovilización, osteogénesis imperfecta, enfermedades inflamatorias y síndrome de Wiedmann-Beckwith.
Una de las causas principales de nefrocalcinosis del lactante es la hipercalciuria debida al uso de furosemida, teofilina y/o corticoides en el prematuro y en el recién nacido con insuficiencia cardiaca(8). Sin embargo, incluso sin estos tratamientos, existe en los grandes prematuros (menos de 1.500 g) una alta incidencia de nefrocalcinosis (del 10 al 60% en función de las series), en cuyo origen destaca la misma prematuridad por la inmadurez tubular y factores nutricionales debidos a la alimentación artificial de estos bebés(9).
En la actualidad, la corticoterapia prolongada en el niño mayor es raramente una causa, así como la sobredosis de vitamina D administrada a título preventivo en el niño sano o a título curativo en enfermedades como la hipofosfatasia, el raquitismo hipofosfatémico ligado al X y la hipocalcemia-hipercalciuria dominante debida a la mutación activadora del receptor sensible al calcio (CaSR)(10).
Hipercalciuria idiopática
La “hipercalciuria idiopática” es una anomalía metabólica de origen genético o familiar, que puede heredarse de modo autosómico dominante, caracterizada por una excesiva eliminación urinaria de calcio en ausencia de hipercalcemia o de otras causas conocidas de hipercalciuria. Este defecto está presente hasta en el 5-10% de la población general, y más frecuentemente en sujetos con litiasis cálcica o población adulta con osteoporosis. Hallazgos obtenidos de trastornos monogénicos caracterizados por cálculos cálcicos y/o hipercalciuria y/o nefrocalcinosis han permitido identificar un número de genes candidatos a participar en la patogénesis de la hipercalciuria idiopática como, por ejemplo: adenilato ciclasa soluble, receptor sensor de calcio, receptor de vitamina D y alfa 1-hidroxilasa, co-transportador 2 sodio-fosfato, claudina 16, canal de cloro-5, etc. Los hallazgos genéticos no apoyan la idea de la existencia de diferentes subtipos de hipercalciuria idiopática clásicos, definidos como: absortivos, renales y resortivos. Al contrario, sugieren que la hipercalciuria idiopática es un único trastorno caracterizado por alteraciones en el transporte de calcio a nivel intestinal, renal y del hueso, debido a diferentes combinaciones de múltiples factores genéticos y dietéticos(11).
Diagnóstico
La determinación en una muestra doble de orina que incluya la primera orina de la mañana y una muestra de orina post-prandial, puede dar suficiente información para identificar una hipercalciuria desde atención primaria. Si únicamente se dispone de muestras aleatorias, sería deseable recoger una muestra recogida tras 2-4 horas de una ingesta láctea. En dicha muestra si la relación Ca/Cr (mg/mg) es menor de 0,2, no sería necesaria una reevaluación de la hipercalciuria. De forma general, los test de sobrecarga de calcio no se recomiendan en la evaluación rutinaria de los niños con hipercalciuria.
El diagnóstico se establece ante una hipercalciuria aislada, en la que se han eliminado posibles causas secundarias, y en la que suelen existir antecedentes familiares de litiasis o nefrocalcinosis. Esta anomalía casi siempre es asintomática en la infancia, hasta su descubrimiento en la edad adulta por la litiasis. Con el tiempo, se observa a menudo una disminución de la densidad ósea, sobre todo cuando está asociada una hipocitraturia(7). Esta forma de hipercalciuria “idiopática” es la causa más frecuente de litiasis renal, tanto en la edad pediátrica como en la adulta, encontrándose en algunas series hasta en el 40% de los niños y en el 60% de los adultos con litiasis(1). Clásicamente, fue considerada una causa mayor de nefrocalcinosis en el niño, representando hasta un tercio de las causas, sin embargo, el descubrimiento de algunas tubulopatías hereditarias, actualmente bien identificadas, ha reducido el número de las hipercalciurias inexplicadas. Por lo tanto, este diagnóstico sólo puede establecerse tras una exploración tubular minuciosa. El conocimiento, cada vez más preciso, de los numerosos genes implicados en el metabolismo del calcio y de la vitamina D permite, de esta forma, desarticular progresivamente la entidad de “hipercalciuria idiopática”, cuyo carácter familiar es bien conocido.
Mecanismo fisiopatológico
La hipótesis más aceptada actualmente es la formulada por Weissinger, que ha postulado una teoría patogénica basada en los hallazgos de varios autores y en los suyos propios, en los que se ha observado un incremento de la actividad de interleucina-1a (IL-1) y de otras citocinas de origen monocitario que incrementarían la actividad osteoclástica, causante de la pérdida de masa ósea observada en estos pacientes(11). La IL-1 estimularía la producción de prostaglandina E2 y ésta, de forma secundaria, el aumento de la producción de calcitriol. La hipercalciuria sería, pues, de origen óseo (resortivo) e intestinal (incremento de la absorción intestinal de calcio). Queda por saber cuáles son las causas que estimulan la producción de citocinas. A pesar de ser un trastorno tan frecuente, aún no se ha descrito ninguna mutación genética responsable única de la hipercalciuria idiopática(12,13).
Manifestaciones clínicas
Referidos a las formas de hipercalciuria aislada o idiopática, la mayor parte de los casos son asintomáticos. En enfermedades en las que la hipercalciuria está presente junto a otras alteraciones, la clínica es variable dependiendo de la enfermedad primaria. Por su frecuencia, la litiasis es la manifestación más relevante, observándose hipercalciuria en un 30-80% de casos según las series.
Otras manifestaciones que pueden presentar los niños, y que permiten sospechar algunas formas de hipercalciuria idiopática, son la presencia de: disuria, polaquiuria, incontinencia urinaria, hematuria macro o microscópica o dolor abdominal recidivante. La hipercalciuria puede aparecer en niños con enuresis nocturna o asociado a infección urinaria recurrente. En ocasiones, sólo se advierte que el niño emite orinas turbias por la presencia de cristales, especialmente, en la primera orina del día. Se cree que los síntomas de la enfermedad se deben a la irritación tisular por la formación de microcristales, aunque se desconoce el mecanismo exacto por el que la hipercalciuria da lugar a hematuria o disuria(14).
Durante los últimos años, se ha evidenciado relación entre hipercalciuria, talla baja y disminución de la densidad mineral ósea(15) y, de forma excepcional, ha sido referido como primer síntoma la aparición de fracturas óseas ante traumatismos de poca intensidad.
Nefrolitiasis
La enfermedad litiásica renal o nefrolitiasis afecta al 3-5% de la población y, a menudo, se asocia a hipercalciuria. La nefrolitiasis hipercalciúrica es una enfermedad familiar que se presenta hasta en el 35% de los pacientes con litiasis, y puede ocurrir como un trastorno monogénico de presentación durante la infancia. Cuando la hipercalciuria predomina, se suele expresar en forma de oxalato de calcio o de fosfato de calcio.
La presencia de cálculos urinarios es una situación poco habitual en la edad pediátrica, pero que condiciona una serie de dilemas clínicos. Especialmente en los más jóvenes, se plantea el dilema de si existe una condición metabólica subyacente, o si existe la posibilidad de que el niño desarrolle, a lo largo de su vida, episodios recurrentes de cólicos nefríticos.
La presentación clínica de dolor cólico agudo es poco habitual en los niños y la sintomatología varía en función de la edad de los niños. Únicamente, la mitad de los niños con urolitiasis manifiestan dolor abdominal, en flancos o pélvico(16,17). En los más pequeños, el dolor puede simular un cólico de lactante. La hematuria, tanto micro como macroscópica, se describe en el 33-90% de los niños con cálculos(16). La hematuria puede preceder a la formación de cálculos en niños con hipercalciuria y ocasionalmente, con hiperoxaluria o hiperuricosuria(18). La urolitiasis puede complicarse con infecciones urinarias, especialmente en los más pequeños, aunque también puede observarse una piuria estéril. Los cálculos en vejiga o uretra con frecuencia se manifiestan con disuria y polaquiuria.
Cuando es posible analizar la composición del cálculo, es posible identificar el origen metabólico que causa la formación de la litiasis. En la edad pediátrica, los cálculos de oxalato de calcio suponen del 46 al 64%, seguidos de los cálculos de fosfato cálcico (14-30%)(16).
Nefrocalcinosis
La nefrocalcinosis se define como la presencia de depósitos de sales de calcio en el parénquima renal, en oposición a la litiasis renal, donde las calcificaciones se sitúan en las vías urinarias. Su frecuencia en el niño, que parecía baja cuando el diagnóstico se basaba en la radiografía de abdomen o en el estudio histológico del riñón, es más importante desde que la ecografía permite su detección precoz, principalmente en los prematuros, en los cuales no es raro detectar una nefrocalcinosis(9).
La nefrocalcinosis no es una enfermedad, sino un síntoma cuyas etiologías son múltiples, pero implican casi todas un trastorno metabólico, a menudo de origen genético o iatrogénico, que aumenta la excreción urinaria del calcio o del oxalato(9,19).
El diagnóstico etiológico está orientado por la edad del niño, el contexto clínico, el aspecto y la localización de las calcificaciones y, sobre todo, por las determinaciones biológicas (al menos un ionograma sanguíneo con determinación de bicarbonato, calcemia, fosfatemia, magnesemia, niveles de 25 OH D3 y 1,25-(OH)2 D3, calciuria y nivel de parathormona, PTH, en caso de hipercalcemia y oxaluria en caso de que la calcemia y la calciuria sean normales) (Fig. 2). En la mayoría de los casos, existe una hipercalciuria, secundaria o no a una hipercalcemia. Más raramente, se trata de depósitos de oxalato de calcio o de la calcificación de lesiones histológicas.
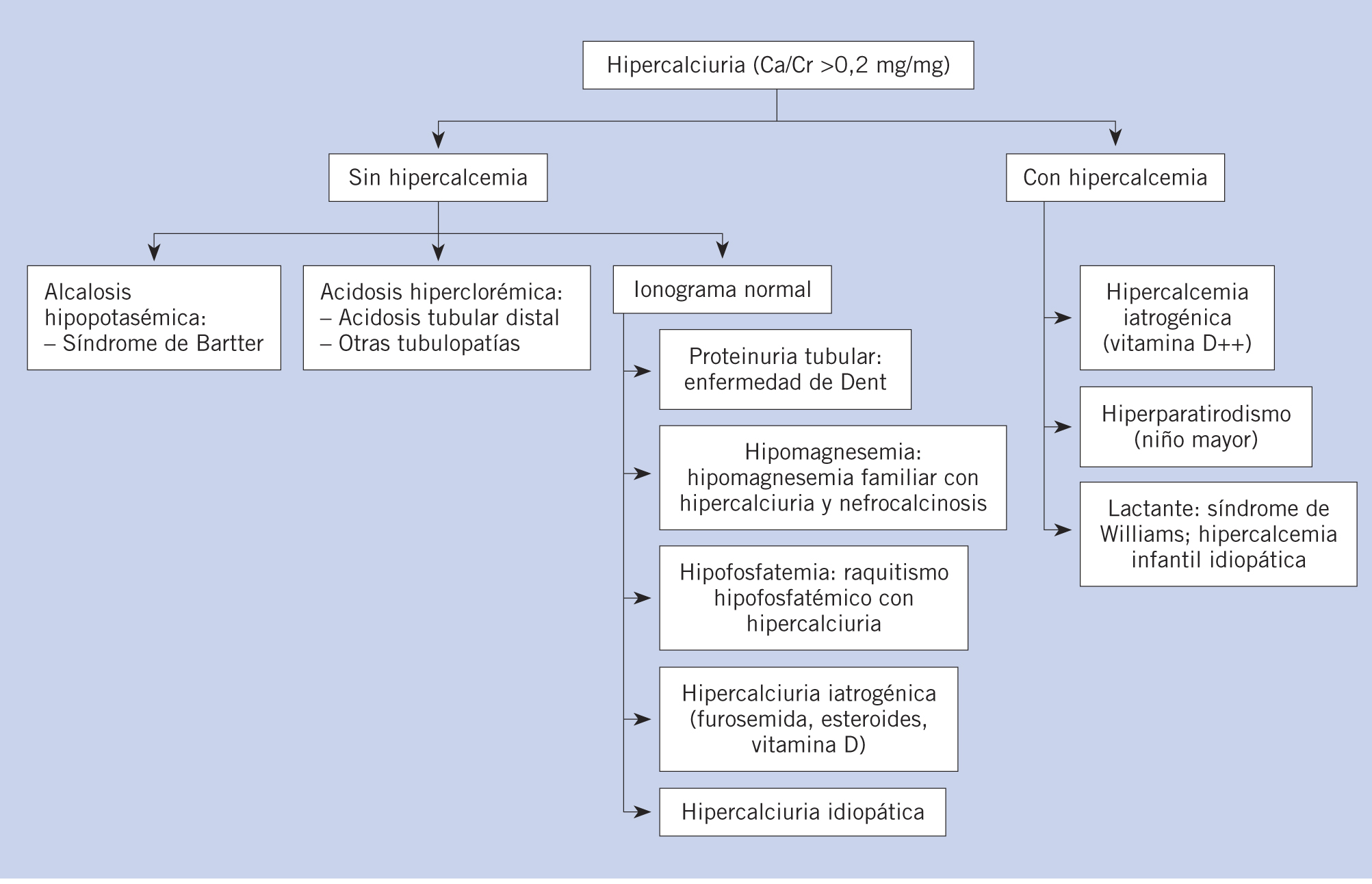
Figura 2. Árbol de decisiones. Conducta práctica ante una hipercalciuria.
La nefrocalcinosis, por sí misma, no suele provocar ningún síntoma clínico antes de llegar a un estadio muy avanzado, que es cuando repercute en la función renal. Casi siempre suele detectarse en una prueba de imagen motivada por los síntomas de una litiasis asociada, o de la enfermedad causal (dolor abdominal, infección urinaria, poliuria, retraso del crecimiento, etc.) o por la existencia de un riesgo conocido (grandes prematuros, ciertas tubulopatías, como el síndrome de Bartter). Los únicos síntomas que pueden imputarse directamente a la nefrocalcinosis son la hematuria, micro o macroscópica, la leucocituria estéril y la alteración de la concentración de la orina.
El diagnóstico suele establecerse cuando las calcificaciones son suficientemente grandes como para ser detectadas en las pruebas de imagen renal. La ecografía renal es la prueba de elección para detectar una nefrocalcinosis. Los depósitos cálcicos se manifiestan como zonas de hiperecogenicidad en el parénquima renal. En función de la localización, se distinguen las nefrocalcinosis medulares, las más frecuentes, en las que existen tres grados según la intensidad, y las nefrocalcinosis corticales o difusas(8,11).
Sin embargo, la hiperecogenicidad no es sinónimo de calcificación (salvo si se acompaña de un cono de sombra acústica) y se deben descartar algunas otras causas de hiperecogenicidad: microquistes de la poliquistosis recesiva, granulomas infecciosos, principalmente por Candida, precipitación intratubular de hematíes en la drepanocitosis (sobre todo, heterocigótica), de uratos en las hiperuricemias congénitas, o de proteína de Tamm-Horsfall en el recién nacido(11). Por otro lado, no hay que olvidar que la ecogenicidad cortical está fisiológicamente aumentada en el recién nacido, lo que puede requerir repetir la prueba al cabo de algunas semanas en caso de duda.
El diagnóstico de nefrocalcinosis debe igualmente confirmarse, primero con una radiografía simple de abdomen. Las calcificaciones pueden ser evidentes o, por el contrario, difícilmente visibles, en función de su tamaño. Son de aspecto variable (en forma de finas semillas, de red, de granos de mijo, grandes bloques opacos), en función de su etiología (Fig. 1). La tomografía computarizada es más sensible y permite detectar calcificaciones no visibles en la radiografía y sus localizaciones precisas, pero su coste y el riesgo ligado a la radiación no permiten su uso como prueba de detección. La resonancia magnética no está indicada en materia de calcificaciones.
Diagnóstico
Anamnesis e historia clínica
Mediante una adecuada historia dietética, debe investigarse la posible existencia de una ingesta excesiva de calcio, proteínas y sodio y, desde el punto de vista clínico, la presencia de dolores óseos o antecedentes de fracturas frecuentes, o las manifestaciones urinarias antes referidas (orinas turbias, disuria, polaquiuria, incontinencia, hematuria, dolores abdominales recidivantes, etc.). Deben recogerse, además, antecedentes familiares de hipercalciuria, de litiasis renal o de consanguinidad y, en la exploración física, considerar una talla ligeramente baja.
Según se recoge en el algoritmo diagnóstico (Figs. 2 y 3), la mayoría de las causas que se identifican como causa de hipercalciuria, y que deben descartarse en todos los casos, son las tubulopatías, alteraciones monogénicas poco frecuentes, en las que suelen existir antecedentes familiares o consanguinidad, fenotipo anómalo, importante retraso de crecimiento, a veces de inicio prenatal, raquitismo, alteraciones hidroelectrolíticas y/o del equilibrio ácido-base, disfunción renal, proteinuria y escasa respuesta al tratamiento (Tabla IV).
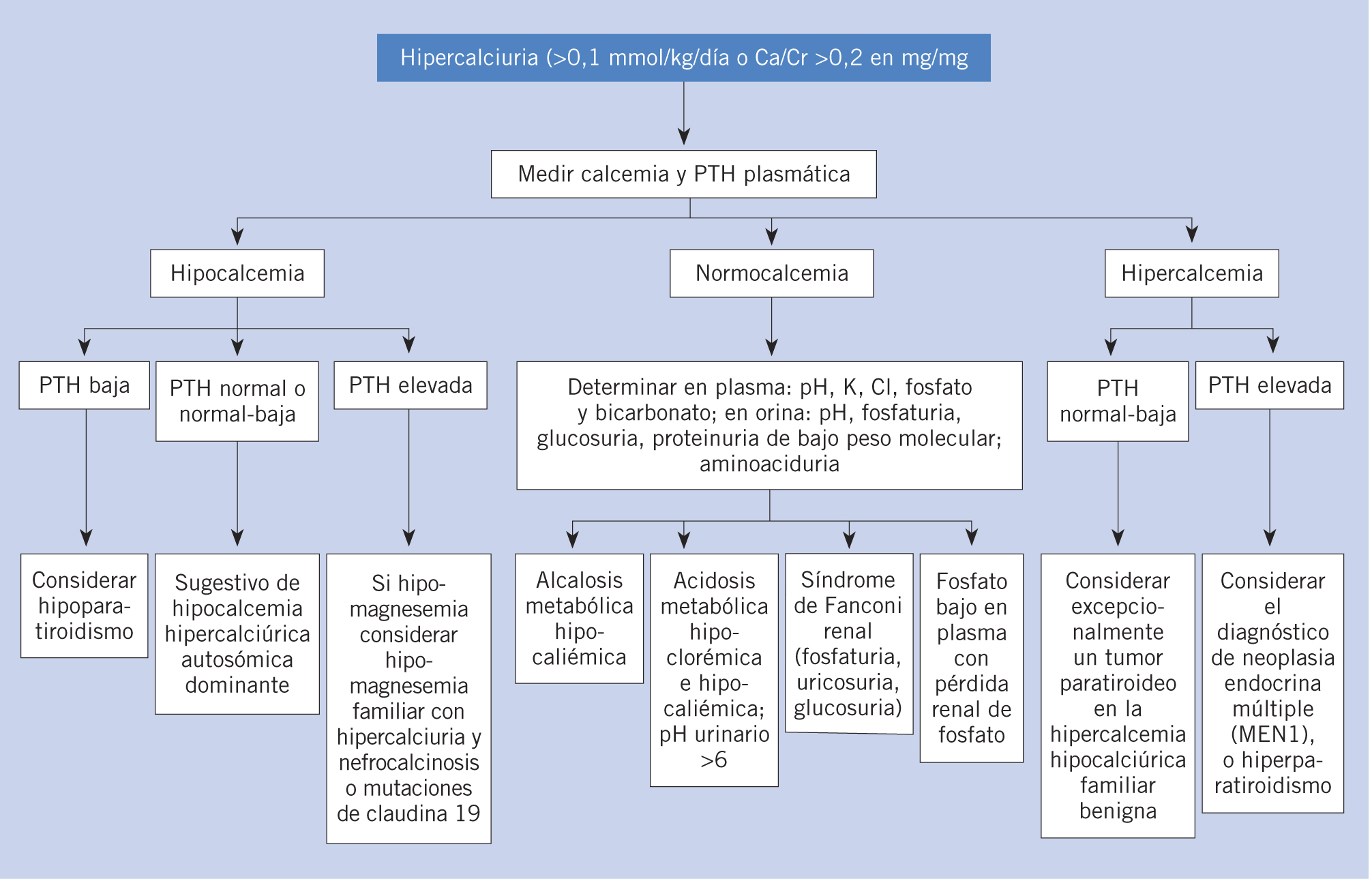
Figura 3. Algoritmo para el estudio de un niño con hipercalciuria y nefrolitiasis/nefrocalcinosis, basado en las determinaciones iniciales de calcemia y PTH plasmática.

Análisis de sangre y orina
Las determinaciones básicas en sangre y orina deben estar dirigidas a descartar otras causas de hipercalciuria.
En todos los niños con hipercalciuria deben determinarse, además de la calciuria, la excreción de citrato urinario y las concentraciones séricas de calcio, fósforo, bicarbonato, magnesio y hormona paratiroidea (PTH). Los estudios diagnósticos para detectar una posible hipercalciuria deben ser diferidos si el paciente tiene una infección urinaria; ya que, durante las pielonefritis, está incrementada la excreción urinaria de calcio.
En primer lugar, debe recogerse una muestra aislada de orina y determinar calcio y creatinina para calcular el cociente Ca/Cr (Tabla I). A partir de los 2-3 años de edad, se concede más valor cuando se trata de la 2ª micción de la mañana, en ayunas. Este cociente debe ser tomado como una aproximación, especialmente adecuada para personas de constitución normal, pero infravalora la calciuria en las muy musculadas. En los niños con escasa actividad muscular, como los portadores de distrofias musculares o mielomeningocele, el cociente sobrevalora la calciuria. Desde el punto de vista del desarrollo, en la pubertad, existe una clara tendencia a descender la eliminación urinaria de calcio.
Antes de realizar el diagnóstico de hipercalciuria, debe confirmarse la hipercalciuria en al menos dos muestras de orina. Una vez identificada la hipercalciuria, es muy importante determinar la citraturia; ya que, en ocasiones, está descendida, con lo que se incrementa el riesgo de litiasis y, quizás, de pérdida de masa ósea. Se considera hipocitraturia los valores inferiores a 8 mg/kg/día o un cociente citrato/creatinina inferior a 400 en mg/g en niños, y a 250 mg/g en adultos (Tabla V).

Una forma sencilla de controlar a los pacientes con hipercalciuria idiopática sería determinar el cociente Ca/Cr en la orina de la noche (al acostarse) y, al día siguiente, en la primera orina del día. El cociente calculado en la orina de la noche estudiaría el componente absortivo (intestinal) y el de la primera orina del día estudiaría el componente resortivo (óseo)(20).
Pruebas de acidificación urinaria
En caso de asociarse hipercalciuria e hipocitraturia, debe realizarse una prueba de acidificación con furosemida. Cuando se objetiva un defecto para descender el pH urinario por debajo de 5,5, debe considerarse la posibilidad de que pueda producirse una evolución hacia el desarrollo de una acidosis tubular distal incompleta.
Ecografía renal y de vías urinarias y otros estudios radiológicos
Permite identificar la existencia de litiasis o de microlitiasis (concreciones de menos de 3 mm de diámetro, sin sombra acústica) y de malformaciones asociadas (muchas malformaciones renales se acompañan de hipercalciuria).
Se solicita una radiografía de mano si la talla es baja. En los casos de fracturas, dolores óseos, hipercalciuria asociada a hipocitraturia, defecto de acidificación tras estímulo con furosemida o presencia de un cociente Ca/Cr elevado en la primera orina del día, está indicado solicitar una densitometría ósea(15). En el caso de encontrarse osteopenia, deben determinarse los niveles de PTH y de los marcadores de remodelado óseo. Los marcadores de formación ósea (función osteoblástica) más utilizados son la osteocalcina y la fosfatasa alcalina ósea. Son marcadores de resorción (función osteoclástica), los niveles de fosfatasa ácida-tartratorresistente y la eliminación urinaria de desoxipiridinolina o de la fracción telopeptídica C-terminal del colágeno en orina. En la mayoría de los casos, la pérdida de masa ósea es de origen resortivo, aunque, en raras ocasiones, existe un defecto en la función osteoblástica.
Tratamiento
Siempre que se identifique una hipercalciuria, el tratamiento dietético debe iniciarse desde Atención Primaria. Una adecuada ingesta de líquidos es crítica en la prevención de la sobresaturación de la orina, independientemente de la causa de la hipercalciuria. Además, está recomendada una dieta pobre en sodio, alta en potasio y baja en oxalatos, para reducir la excreción urinaria de calcio.
En los niños con hipercalciuria, debe garantizarse una adecuada ingesta de líquidos para la prevención de la sobresaturación de la orina, independientemente de la causa. Además, debe recomendarse una dieta pobre en sodio, alta en potasio y baja en oxalatos en los niños con hipercalciuria, oxaliuria y litiasis idiopática, debido al conocido efecto calciúrico de una dieta rica en sal. Una dieta aumentada en potasio puede reducir la excreción urinaria de calcio(21).
Si no se trata, la hipercalciuria conduce a nefrolitiasis en aproximadamente el 15% de los casos. En los casos refractarios, junto a la dieta, la administración de diuréticos tiazídicos por vía oral puede normalizar la excreción urinaria de calcio mediante la estimulación de la reabsorción de calcio en el túbulo contorneado proximal y distal. Dicha terapia detiene la hematuria macroscópica y la disuria y evita el desarrollo de nefrolitiasis. Sin embargo, aún no se han establecido de manera precisa las indicaciones del tratamiento con diuréticos tiazídicos en la hipercalciuria idiopática.
Normas dietéticas
La situación de riesgo que supone una hipercalciuria asintomática exige únicamente la instauración de medidas dietéticas protectoras e incremento de la práctica de ejercicio físico, evitando crear conciencia de enfermedad. La dieta debe ajustarse a las recomendaciones diarias para la edad para la ingesta de proteínas. Debe suspenderse el aporte de suplementos de vitamina C y/o vitamina D.
La situación de riesgo que supone una hipercalciuria asintomática exige únicamente la instauración de medidas dietéticas protectoras e incremento de la actividad física, evitando crear conciencia de enfermedad. En las formas no complicadas debe favorecerse el seguimiento desde Atención Primaria. La dieta debe ajustarse a las recomendaciones diarias para la edad respecto a la ingesta de proteínas, calcio y sal. Debe suspenderse el aporte de suplementos de vitamina C y/o vitamina D.
De forma general, las recomendaciones serían las siguientes:
a. Ingesta abundante de agua: 30 ml/kg/día sin exceder de 2 litros. Lo ideal sería conseguir diuresis superiores a 750 ml en lactantes, a 1.000 ml en niños menores de 5 años, a 1.500 ml en niños de 6 a 10 años y superiores a 2 litros en preadolescentes y adolescentes.
b. Evitar el exceso de calcio, adecuando los aportes a las necesidades, favoreciendo el consumo de frutas en vez de postres lácteos. La restricción de calcio no se recomienda por la asociación de la hipercalciuria con osteopenia, pero deben garantizarse las necesidades diarias de ingesta de calcio, sin que deban sobrepasarse, a pesar de los estímulos culturales hacia la ingesta de productos lácteos. La recomendación general es no exceder de 500 ml al día de lácteos.
La mayoría de leches comerciales contienen 120 mg/dl. De estos aportes se absorbe, a nivel intestinal, aproximadamente, el 40%, excepto cuando se trata de leche materna que alcanza el 60%.
c. Dado que la calciuria aumenta al hacerlo la natriuria, y disminuye cuando ésta lo hace o aumenta la kaliuria, está indicada una restricción relativa de sodio y favorecer el consumo de alimentos ricos en potasio. Entre los alimentos de consumo habitual que presentan mejor relación, podemos subrayar: legumbres (habas y alcachofas), verduras y hortalizas (tomates y berenjenas), frutas (plátano, uva, fresón, melón, cerezas, ciruela, níspero, higo y piña) y frutos secos (almendras, cacahuetes, avellanas y castañas). La monitorización de la natriuresis es de utilidad para evaluar el cumplimiento de las recomendaciones dietéticas. Las recomendaciones de ingesta de sal para los menores de 3 años serían de menos de 1,5 g/día, de 4 a 8 años menos de 1,9 g, de 9 a 13 años menos de 2,2 g y de 14 a 18 años menos de 2,3 g.
d. Aporte de citrato en la dieta. Los cítricos (naranja, limón) tienen un alto contenido en citrato, que es el principal inhibidor de la cristalización. En segundo lugar, la excreción urinaria de potasio disminuye la calciuria. Muchas frutas son además ricas en potasio (sandía, melón, naranja, mandarina, uvas, plátano).
e. Restricción del aporte de proteínas animales, ya que éstas incrementan la calciuria al elevar la excreción neta de ácido, al mismo tiempo que aumentan la eliminación de ácido úrico y de oxalato. La alternativa que suponen los cereales integrales y el pescado azul tiene la ventaja de aportar factores protectores de la formación de cálculos.
En la tabla VI, aparece un resumen de las normas dietéticas que puede ser entregado a las familias de los niños afectos(22).

Tratamiento farmacológico
1. En las hipercalciurias sintomáticas importantes o con riesgo de complicaciones, que no respondan a las medidas dietéticas y, más especialmente, en los niños con excreción reducida de citrato, se añade al tratamiento citrato potásico a dosis de 1-1,5 mEq/kg/24 horas. El citrato en la orina forma complejos con el calcio, reduciendo el calcio iónico y la formación de sales de oxalato y de fosfato cálcicos. Un aspecto adicional es que el citrato es una fuente de energía para el riñón y es un componente óseo (supone el 1% de su peso), pudiendo estar especialmente indicado en los niños con osteopenia.
2. En caso de no tolerarse el citrato o de no existir mejoría, se añade hidroclorotiazida: 1,5-2,5 mg/kg en dosis única matutina(23). Las tiazidas estimulan el transporte transcelular de calcio en el túbulo contorneado distal. La dosis se va incrementando hasta conseguir una excreción de calcio en orina de 24 horas menor de 4 mg/kg y la desaparición de las manifestaciones clínicas, si las hubiese. El tratamiento se mantiene un año, tras el cual, en el caso de las hipercalciurias idiopáticas, puede suspenderse, pero deberá reiniciarse en caso de recidiva de la hematuria macroscópica, de la nefrolitiasis o de la disuria.
Hay que tener en cuenta que el tratamiento con diuréticos tiazídicos puede tener efectos secundarios, lo cual obliga a realizar controles periódicos plasmáticos. El más frecuente e importante es la hipopotasemia, que puede evitarse asociando amiloride. Además, pueden favorecer hipomagnesemia, aumento de los niveles plasmáticos de glucosa, urato, colesterol y LDL, y reducir la eliminación urinaria de citrato.
3. En caso de osteopenia severa, se está estudiando el efecto antirresortivo de los bifosfonatos, cuya utilidad ha sido probada en adultos y niños hipercalciúricos con desmineralización ósea y en otras patologías pediátricas(24).
Bibliografía
Los asteriscos reflejan el interés del artículo a juicio del autor.
1.** Milliner DA. Urolithiasis. En: Avner ED, Harmon WE, Niaudet P, eds. Pediatric Nephrology. 5ª ed. Philadelphia: Lippincott Willians & Wilkins; 2004. p. 1091-111.
2.* Sargent JD, Stukel TA, Kresel J, et al. Normal values for random urinary calcium to creatinine ratios in infancy. J Pediatr. 1993; 123: 393-7.
3.** Kruse K, Kracht U, Kruse U. Reference values for urinary calcium excretion and screening for hypercalciuria in children and adolescents. Eur J Pediatr. 1984; 143: 25-31.
4. Stapleton FB, Noe HN, Jerkins GR, et al. Urinary excretion of calcium following an oral calcium loading test in healthy children. Pediatrics. 1982; 69: 594-7.
5. Theodoropoulos DS, Shawker TH, Heinrichs C, Gahl W. Medullary nephrocalcinosis in nephropathic cystinosis Pediatr Nephrol. 1995; 9: 412-8.
6.* Cochat P, Pichault V, Bacchetta J, Dubourg L, Sabot C, Daudon M, et al. Nephrolithiasis related to inborn metabolic diseases. Pediatr Nephrol. 2010; 25: 415-24.
7. Vargas-Poussou R, Cochat P, Le Pottier N, Roncelin I, Liutkus A, Blanchard A, et al. Report of a family with two different diseases leading to early nephrocalcinosis Pediatr Nephrol. 2008; 23: 149-53.
8. Saarela T, Vaarala A, Lanning P, Koivisto M. Incidence, ultrasonic patterns and resolution of nephrocalcinosis in very low birth weight infants. Acta Paediatr. 1999; 88: 655-60.
9.* Schell-Feith EA, Kist-van-Holthe JE, van der Heijden AJ. Nephrocalcinosis in preterm neonates. Pediatr Nephrol. 2010; 25: 221-30.
10. Auron A, Alon US. Resolution of medullary nephrocalcinosis in children with metabolic bone disorders. Pediatr Nephrol. 2005; 20: 1143-5.
11.** Weisinger JR. New insights into the pathogenesis of idiopathic hypercalciuria: The role of bone. Kidney Int. 1996; 49: 1507-18.
12.*** Vezzoli G, Soldati L, Gambaro G. Update on primary hypercalciuria from a genetic perspective. J Urol. 2008; 179(5): 1676-82.
13.* Stechman MJ, Loh NY, Thakker RV. Genetic causes of hypercalciuric nephrolithiasis. Pediatr Nephrol. 2009; 24(12): 2321-32.
14. Alon U, Warady BA, Hellerstein S. Hypercalciuria in the frequency-dysuria syndrome of childhood. J Pediatr. 1990; 116: 103-5.
15. García-Nieto V, Ferrández C, Monge M, de Sequera M, Rodrigo MD. Bone mineral density in pediatric patients with idiopathic hypercalciuria. Pediatr Nephrol. 1997; 11: 578-83.
16. Milliner DS, Murphy ME. Urolithiasis in Pediatric Patients. Mayo Clin Proc. 1993; 68: 241-8.
17. Stapleton FB, Roy S, Noe HN. Hypercalciuria in children with hematuria. N Engl J Med. 1984; 310: 1345-8.
18. Voghenzi A, Bezzi TM, Luscardi P, et al. Acquired hyperoxaluria and haematuria in children. Pediat Nephrol. 1992; 6: 356-7.
19. Ammenti A, Pelizzoni A, Cecconi M, Molinari PP, Montini G. Nephrocalcinosis in children: a retrospective multi-centre study. Acta Paediatr. 2009; 98: 1628-31.
20. García-Nieto V, Monge Zamorano M, Rodrigo Jiménez MD, Callejón Callejón A, Gaspar Guardado A, García Rodríguez VE. Estudio evolutivo de la densidad mineral ósea en niños diagnosticados de hipercalciuria idiopática. Su relación con la calciuria y con la eliminación urinaria de desoxipiridinolina, determinadas en dos momentos del día. Nefrología. 2004; 24: 610.
21.* Osorio AV, Alon US. The relationship between urinary calcium, sodium and potassium excretion and the role of potassium in treating idiopathic hypercalciuria. Pediatrics. 1997; 100: 675-81.
22.** Fons J, García-Nieto V. Hipercalciuria idiopática. En: Protocolos Diagnóstico Terapéuticos de la AEP: Nefrología Pediátrica. Asociación Española de Pediatría; 2008. p. 182-8.
23.* Srivastava T, Alon US. Pathophysiology of hypercalciuria in children. Pediatr Nephrol. 2007; 22: 1659-73.
24. Barrios E, Hernández González MJ, Rodrigo Jiménez MD, Armas Suárez S, Claverie-Martín F, García-Nieto V. Efectiveness of treatment with bisphosphonates in childhood osteoporosis. Pediatr Nephrol. 2006, 21: 1588.
Bibliografía recomendada
– Milliner DA. Urolithiasis. En: Avner ED, Harmon WE, Niaudet P, eds Pediatric Nephrology. 5ª ed. Philadelphia: Lippincott Willians & Wilkins; 2004. p. 1091-111.
Trabajo clásico de fácil lectura en un texto de referencia en nefrología pediátrica. Desde el problema de la urolitiasis, se acerca a la hipercalciuria como condición metabólica relevante en pediatría. Se recogen todos los aspectos clínicos y terapéuticos relevantes.
– Vezzoli G, Soldati L, Gambaro G. Update on primary hypercalciuria from a genetic perspective. J Urol. 2008; 179(5): 1676-82.
Excelente revisión sobre los recientes hallazgos genéticos en la hipercacluria primaria. Se consideran aspectos patogénicos y su implicación con las posibles complicaciones de esta alteración metabólica asociada a trastornos del transporte del calcio en el intestino, riñón y hueso. Se documenta la elevada prevalencia de esta condición y las posibles manifestaciones en la edad pediátrica.
– Weisinger JR. New insights into the pathogenesis of idiopathic hypercalciuria: The role of bone. Kidney Int. 1996; 49: 1507-18.
Excelente trabajo para los lectores interesados en profundizar en las bases patogénicas de esta alteración. Especial interés en el metabolismo óseo.
– Stechman MJ, Loh NY, Thakker RV. Genetic causes of hypercalciuric nephrolithiasis. Pediatr Nephrol. 2009; 24(12): 2321-32.
Estos autores proponen un algoritmo de utilidad clínica para la investigación de las causas genéticas de nefrolitiasis a partir del hallazgo de una hipercalciuria.
– Kruse K, Kracht U, Kruse U. Reference values for urinary calcium excretion and screening for hypercalciuria in children and adolescents. Eur J Pediatr. 1984; 143: 25-31.
Trabajo clásico sobre valores de referencia útiles en pediatría y aplicables en múltiples estudios posteriores. Este trabajo dimensiona la magnitud de la hipercalciuria y la utilidad del cribado de esta condición.
– Fons J, García-Nieto V. Hipercalciuria idiopática. En: Protocolos Diagnóstico Terapéuticos de la AEP: Nefrología Pediátrica. Asociación Española de Pediatría 2008. p. 182-8.
Práctica publicación sobre el manejo clínico en Atención Primaria de las hipercalciurias idiopáticas, con especial hincapié en las recomendaciones dietéticas. Los autores tienen una amplia experiencia en las complicaciones de esta entidad y en la necesidad de realizar un abordaje dietético precoz asumible por el paciente.
| Caso clínico. |
|
Niña de 5 años de edad que consulta por disuria asociada a microhematuria. El cuadro clínico se inició un mes antes, con leves molestias urinarias y sin otros síntomas asociados. En el primer examen de orina realizado, en la tira de orina se detectó la presencia de microhematuria sin otras alteraciones, siendo el urocultivo negativo. En la entrevista clínica dirigida, la niña se queja únicamente de discreto dolor para orinar y en la exploración física no se detectan alteraciones significativas, con ausencia de edemas y peso, talla y tensiones arteriales normales para su edad. La familia niega la presencia de fiebre asociada al episodio, polaquiuria, hematuria, dolor lumbar o abdominal, y tampoco refieren historia de traumatismo ni episodios similares previos. Entre los antecedentes familiares, destaca una historia de litiasis en línea paterna (padre y abuelo). No se recogen otros antecedentes de interés. Ante la presencia de microhematuria, se solicita un sedimento urinario con estudio de la morfología de hematíes y una ecografía renal. En el sedimento urinario se detecta la presencia de hematuria microscópica (40.000 hematíes/mm3), sin proteinuria, dimorfismo de los hematíes ni otras alteraciones. Los estudios ecográficos de las vías urinarias son normales. Se solicita calcio (Ca) y creatinina (Cr) en una muestra aislada de orina para el cálculo de la relación Ca/Cr. La determinación en la primera muestra de orina, obtenida en ayunas, demuestra una relación de 0,24 mg/mg (valores normales <0,2), y en una segunda, postprandial, de 0,32. Se recoge orina de 24 horas para la estimación de la calciuria, que muestra un valor de 5,6 mg/kg/24 h (normal <4 mg/kg/24 horas), lo que confirma el diagnóstico de hipercalciuria. Otros exámenes realizados en sangre fueron los siguientes: gasometría (normal); urea, creatinina y ácido úrico (normales), electrolitos, incluyendo Na, K, Ca, P, Mg (normales); y PTH, normal. En orina, se solicita pH (6,5), natriuresis (200 mEq/L), oxaluria (normal), citraturia (disminuida) y uricosuria (elevada). Con el diagnóstico de hipercalciuria asociada a uricosuria se orienta a la familia acerca de la necesidad de aumentar la ingesta hídrica, restringir la ingesta de sodio en la dieta y ajustar la ingesta de lácteos a las recomendaciones para su edad. Durante el seguimiento, se monitoriza la natriuresis para evaluar la adherencia a las recomendaciones en relación a la ingesta de sodio. El primer control de sodio en orina fue de 189 mEq/L y el siguiente de 80 mEq/L. Se programa seguimiento ambulatorio con ecografía renal cada 12 meses y controles clínicos semestrales, incluyendo calciuria y natriuria. |
