 |
| Temas de FC |
C. Gutiérrez Segura, Á. Gómez Farpón, C. Granell Suárez
Médicos Adjuntos (FEA) del Servicio de Cirugía Pediátrica.
Hospital Universitario Central de Asturias
| Resumen
Las anomalías congénitas del riñón y del tracto urinario (CAKUT) representan aproximadamente el 20-30% de todas las anomalías identificadas en el periodo neonatal, dando lugar a una amplia variedad de desórdenes. |
| Abstract
Congenital anomalies of the kidney and urinary tract (CAKUT) account for approximately 20-30% of all abnormalities identified in the neonatal period resulting in a wide variety of disorders. |
Palabras clave: Hipoplasia y displasia renal; Riñón multiquístico; Ectopia renal; Riñón en herradura; Estenosis pielo- ureteral; Duplicidad renal y ureteral; Megauréter; Uréter ectópico; Ureterocele; Válvulas de uretra; Reflujo vésico-ureteral.
Key words: Renal hipoplasia and displasia; Renal ectopia; Horseshoe Kidney; Multicystic dysplastic Kidney; Urete- ropelvic junction obstruction; Duplicate collecting systems; Megaureter; Ectopic uréter; Ureterocele; Posterior urethral valve; Vesicoureteral reflux.
Pediatr Integral 2013; XVII(6): 391-401
Anomalías congénitas del riñón y del tracto urinario
Anomalías congénitas del riñón y del tracto urinario (CAKUT)
Las CAKUT constituyen cerca del 20-30% de todas las malformaciones identificadas(1), ocurriendo en el 0,3-1,6/1.000 recién nacidos vivos (RNV), ocasionando un 30-50% de insuficiencia renal terminal (IRT), siendo importante su diagnóstico y tratamiento precoz para minimizar el daño renal.
Representan una amplia variedad de desórdenes que provienen de los siguientes procesos de desarrollo renal anormal:
a. Malformación del parénquima renal: hipoplasia, displasia, agenesia renal y riñón multiquístico (DRMQ).
b. Anomalías relacionadas con la migración de los riñones: ectopia renal y anomalías de fusión.
c. Anomalías en el desarrollo del sistema colector: estenosis pielo-ureteral (EPU), duplicidades ureterales, megauréter primario, uréter ectópico, ureterocele y válvulas de uretra posterior (VUP).
Revisaremos aquellos que tienen un mayor impacto y repercusión clínica desde el punto de vista quirúrgico.
Malformaciones del parénquima renal
Su patogénesis es multifactorial, siendo debidas a factores genéticos y ambientales. En modelos animales, se ha demostrado cómo la deficiencia de vitamina A está asociada con malformaciones urogenitales e hipoplasia renal.
Hipoplasia renal
Existe un bajo número de nefronas, estructuralmente normales (sin cicatrices), dando lugar a un riñón disminuido de tamaño y una hipertrofia del riñón contralateral.
Displasia renal
Caracterizada por la presencia de tejido renal malformado, puede ser uni o bilateral, ocurriendo en 2-4/1.000 RNV. El diagnóstico frecuentemente es casual durante la realización de una ecografía. Suelen ser de tamaño más pequeño y su evolución, en general, es buena, en casos unilaterales.
Debido a la frecuente asociación con otras anomalías del sistema colector, la cistouretrografía miccional seriada (CUMS) debería ser considerada en todos los pacientes con displasia renal. La gammagrafía isotópica (DMSA) puede proporcionar información adicional de la función renal diferencial (FRD) de cada riñón. Se deben realizar ecografías periódicas para controlar el crecimiento compensatorio contralateral y posibles cambios en la medida del riñón displásico.
Displasia renal multiquística (DRMQ)
Es un riñón no funcionante, con múltiples quistes no comunicantes separados por tejido displásico con un uréter ausente o atrésico. La incidencia global es de 0,3-1/1.000 RNV, con más de la mitad de los casos diagnosticados antes del nacimiento. Suele existir involución o desaparición del riñón multiquístico en el 90% de los casos. Debido a ello, se recomienda tratamiento conservador con seguimiento a largo plazo.
La mayoría son unilaterales, afectando más frecuentemente a varones en el riñón izquierdo con hipertrofia compensadora en el riñón contralateral sano. Se asocia con anomalías genitales, renales o ureterales contralaterales, siendo el reflujo vésico-ureteral (RVU) lo más frecuente (25% de los casos)(2).
Seguimiento. Evaluación ecográfica seriada hasta los 10 años para monitorizar el crecimiento del riñón contralateral y la involución del DRMQ. Exámenes rutinarios de toma de presión arterial, análisis de orina para detectar proteinuria y estudios de función renal, especialmente en niños con anomalías en el riñón contralateral. Si hubiera historia de infecciones del tracto urinario (ITU) o hidronefrosis (HN), hay que realizar CUMS para diagnosticar la existencia del RVU.
Tratamiento. Algunos clínicos han recomendado eliminar el riesgo de malignidad (tumor de Wilms), extirpando el DRMQ. Sin embargo, en una revisión de la literatura realizada sobre un total de 1.041 niños con riñón multiquístico, ninguno desarrolló tumor de Wilms(3).
Anomalías de la migración y de la fusión
El crecimiento caudal rápido del embrión da lugar a la migración del riñón en desarrollo desde la pelvis a la fosa renal retroperitoneal. Al tiempo que ambos riñones ascienden, rotan 90°, de manera que el hilio renal se dirige hacia medial una vez completada su formación hacia la 8ª semana de gestación.
Ectopia renal
Ocurre cuando el riñón no asciende normalmente para alcanzar la fosa renal. Su incidencia es de 1/5.000 RNV. La mayoría son asintomáticos. El riñón ectópico suele tener una FRD disminuida. La anomalía acompañante más frecuente es el RVU (20%). En ausencia de otras anomalías, el pronóstico de los pacientes con ectopia renal es bueno.
La ectopia puede ser simple, cuando el riñón alcanza una posición anómala pero en el lado correspondiente, y cruzada, cuando cruza la línea media ocupando el lado contralateral. Ésta puede ocurrir con o sin fusión con el riñón contralateral. Si el riñón no asciende se denomina riñón pélvico. En los riñones ectópicos falla la rotación, lo que ocasiona desviación del eje renal, quedando la pelvis renal dirigida anteriormente, siendo el aporte vascular variable y anómalo.
Clínica. Los casos sintomáticos son debidos a ITU, obstrucción o cálculos renales. Pueden existir anomalías acompañantes génito-urinarias o asociación con síndromes, como CHARGE y el de VACTERL.
Diagnóstico. La ecografía postnatal confirma los hallazgos prenatales y define la anatomía. Deben realizarse estudios de función renal en casos de enfermedad renal bilateral o hidronefrosis. Si la función renal está disminuida o si el riñón contralateral es anormal, se debe realizar DMSA para medir la FRD. Si existe HN y la CUMS es normal, entonces se debe realizar MAG-3 para detectar obstrucción.
Fusión renal
Ocurre cuando una parte de un riñón se fusiona con el otro riñón.
Riñón en herradura
Es la anomalía de fusión más frecuente, ocurriendo en 0,4-1,6/10.000 RNV. Un tercio a mitad de los pacientes tienen anomalías congénitas urológicas y genitales acompañantes, así como alteraciones genéticas. Una gran parte de los pacientes tienen un buen pronóstico; sin embargo, estos niños parecen tener un riesgo incrementado de desarrollar tumor de Wilms(4).
En más del 90% de los casos, la fusión ocurre en los polos distales, manteniendo dos sistemas excretores renales y ureterales separados. La porción fusionada o istmo puede estar formado por parénquima o tejido fibroso en la línea media. Los ejes renales están desviados, situándose generalmente las pelvis renales anteriormente, cruzando ambos uréteres por encima del istmo renal fusionado.
Clínica. La mayoría son asintomáticos, aunque la hidronefrosis ocurre en un 80% de los casos, siendo las causas un RVU u obstrucción del sistema colector por compresión ureteral debida a vasos aberrantes, cálculos renales (20%) o por EPU, debido a la inserción alta de los uréteres en la pelvis renal.
Diagnóstico. La ecografía define la alteración anatómica y permite apreciar la HN. Si hay historia de ITU, se debe realizar CUMS para determinar si existe RVU. La creatinina permite evaluar la función renal. Si está elevada, se debe realizar DMSA para medir la FRD y descartar cicatrices renales. En pacientes con HN no debida a RVU debe realizarse MAG-3 para estudiar la EPU.
Tratamiento. El riñón en herradura debe ser intervenido solamente cuando en él se desarrolle un proceso patológico que evolucione desfavorablemente. A la hora de operar un RVU o una HN, es útil tener un mapa arteriográfico, debido a la vascularización anárquica que presenta el riñón en herradura.
Anomalías en el desarrollo del sistema colector
Estenosis pieloureteral (EPU)
La estenosis entre la pelvis renal y el uréter produce una interrupción parcial o total al flujo de orina, lo que conlleva una HN y, en ciertos casos, una pérdida progresiva de función renal. No obstante, la HN es un signo radiológico, no siempre sinónimo de obstrucción, por lo que, ante una dilatación renal, debemos descartar otras causas (Tabla I).

Epidemiología. Es la causa más común de HN prenatal, aunque muchas de éstas desaparecen. 1/500 fetos tienen una EPU funcionalmente significativa. Es 2 veces más frecuente en el lado izquierdo, predomina ligeramente en varones y en el 10-40% es bilateral.
Etiología. Las EPU pediátricas son fundamentalmente congénitas, pudiendo encontrar algún caso adquirido secundario a cálculos, tumores o cirugías. A su vez, las causas congénitas pueden dividirse en: intrínsecas, las más frecuentes, producidas por un segmento ureteral adinámico e hipoplásico; y extrínsecas, debido a factores mecánicos que comprimen el uréter proximal, como vasos polares, angulaciones de la unión o pólipos.
Clínica. Desde la introducción del screening prenatal, menos del 20% de los casos se diagnostican por su clínica. Cuando esto sucede, lo más frecuente es tras un episodio de ITU febril y, en menor medida, por hematuria tras un traumatismo banal, dolor abdominal, como un hallazgo incidental o formando parte de un síndrome, como en el VACTERL. La crisis de Dietl es típica de escolares mayores, con episodios de vómitos y dolor abdominal cólico asociados a hidronefrosis intermitente secundaria a vasos polares.
Diagnóstico:
• Ecografía: útil para el seguimiento del paciente. No puede diagnosticar la obstrucción per se; ya que, la dilatación severa no es específica de la obstrucción. Debemos prestar atención al grado de dilatación pielocalicial, el tamaño renal, el adelgazamiento cortical, la ecogenicidad cortical, la dilatación ureteral, el grosor vesical y la orina residual. Dos son los sistemas de clasificación del grado de HN: el diámetro antero-posterior de la pelvis (DAP), medida fácilmente reproducible, y el propuesto por la Society for Fetal Urology (SFU). Durante la etapa prenatal, se acepta como HN un DAP >4 mm antes de las 33 semanas de gestación y >7 mm después de esa semana. En la etapa posnatal, toda HN diagnosticada prenatalmente debe estudiarse una vez transcurridos unos 5 días de vida, evitando al menos las primeras 48 horas donde la oliguria fisiológica puede darnos un falso negativo. Sólo en casos seleccionados, como una HN de alto grado, casos bilaterales en varones con el fin de descartar VUP, HN en pacientes monorrenos u oligohidramnios, debe efectuarse antes.
• CUMS: su indicación es controvertida, al ser una prueba invasiva. A su favor está el hecho de que, en la bibliografía, figura una prevalencia de RVU del 25% en HN postnatales, sin embargo, este RVU podría carecer de repercusión clínica y no modificar nuestra actitud terapéutica.
• Renograma diurético MAG-3: es la principal herramienta diagnóstica de obstrucción del tracto urinario, valorando la FRD, que es la proporción de trazador captado de la sangre por cada riñón, y la excreción renal. Debe realizarse transcurrido el mes de vida, cuando el sistema renal es más maduro.
Durante el estudio, se administra un diurético (furosemida), generalmente a los 20 minutos de la máxima actividad del radiotrazador. La valoración del renograma incluye tres aspectos: la visualización de las imágenes secuenciales, las curvas de captación-eliminación (Fig. 1) y los datos cuantitativos, destacando la FRD y el T1/2 (tiempo medio de eliminación)(5). Se considera una FRD deteriorada cuando es <40%. En este punto, es básico recordar que la FRD es un estudio comparativo, es decir, se asume que conjuntamente ambos sistemas renales poseen una función total del 100%, viendo cuánto contribuye cada uno. Por lo tanto, en pacientes monorrenos o con afectación bilateral, este parámetro no es útil. Respecto al T1/2 a los 10 y 20 minutos, o tiempo que tarda en eliminar el 50% del radiofármaco, diferenciamos distintos patrones renográficos. De este modo, un T1/2 <10 minutos es normal (no obstructivo), >20 minutos está alterado (generalmente, asociado con obstrucción) y entre 10 y 20 minutos se considera indeterminado(6). Para lograr un diagnóstico preciso, es esencial prestar atención a múltiples factores que pueden alterar el resultado y producir una curva falsamente obstructiva. Por ello, un drenaje alterado en el renograma no es sinónimo de obstrucción. Sin embargo, una eliminación adecuada implica la ausencia de obstrucción.
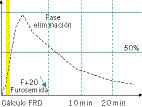
Figura 1. Curva de captación-eliminación en el MAG-3.
• DMSA: proporciona una evaluación más exacta de la FRD que el MAG-3, especialmente en pacientes con riñones poco funcionantes, aunque con una dosis de radiación superior.
• Otras pruebas: en desuso han quedado los estudios de presión-flujo o la UIV, en detrimento de pruebas más precisas, como la urografía por RMN y el TC con contraste.
• Criterios de obstrucción y cirugía: las indicaciones de cirugía se encuentran en continuo debate. Por un lado, no hay ningún parámetro que, por sí solo, pueda indicarnos tratamiento quirúrgico, y la mera observación de la curva de eliminación es un manejo simplista de un problema muy complejo. Por otro lado y, gracias al uso de la ecografía prenatal, se sabe que la mayoría de las HN (75%) diagnosticadas en este periodo permanecen estables o mejoran, sin poder determinar los factores de deterioro renal y pronosticar qué pacientes no necesitarán cirugía. En general, la mayoría de las HN con DAP >30 mm precisarán cirugía (90%) frente al 10% con pelvis <20 mm. En los pacientes con pelvis de 20-30 mm, la mejoría (60%) es casi tan probable como el deterioro. Los criterios de cirugía son la presencia de síntomas, descenso de la FRD o incremento de la HN. En casos dudosos, debemos seguir regularmente al paciente con ecografía y MAG-3(7).
Tratamiento:
a. Pieloplastia: la pieloplastia desmembrada de Anderson-Hynes sigue considerándose el tratamiento de elección: se reseca la estenosis y parte de la pelvis renal redundante y se anastomosan pelvis y uréter. Esta técnica puede llevarse a cabo por cirugía abierta, laparoscopia o robótica.
b. Endourología: este tipo de procedimientos técnicos mínimamente invasivos están ampliamente instaurados en el adulto por su simplicidad e inocuidad. Sin embargo, en el paciente pediátrico tienen unos resultados muy discutidos y las dificultades técnicas hace que se lleven a cabo en centros muy concretos. Se puede acceder a la zona estenótica a través de la uretra y del uréter (retrógradamente) o a través de la pelvis renal (anterógradamente) y, con diferentes dispositivos, conseguir vencer esta estenosis (balón de dilatación de alta presión, endopielotomía…)(8).
Seguimiento postoperatorio. La mejoría del lavado del trazador en el MAG-3 después de la pieloplastia es muy variable, pudiendo, en ocasiones, permanecer obstruido o equívoco al menos durante 6 meses. Si la FRD permanece estable, no hay necesidad de preocuparse.
Megauréter congénito
La definición de “megauréter” es puramente descriptiva: es aquel uréter con un diámetro superior a 7 mm, acompañado o no de dilatación del sistema colector alto y sin hacer referencia a la obstrucción o el RVU.
Se diferencian 4 tipos con diferente etiopatogenia, en función de que sea obstructivo y/o refluyente:
1. Obstruido no refluyente o megauréter primario obstructivo (MOP), secundario a una obstrucción parcial por una alteración en la musculatura ureteral distal(9). Aunque, para otros autores, el hecho de que algunos se resuelvan de forma espontánea hace pensar que sea una falta de maduración del uréter(10).
2. Obstruido y refluyente, en el cual el trayecto tunelizado urétero-vesical es anormal y la orina refluye al uréter pero es incapaz de volver a la vejiga.
3. No obstruido refluyente, donde la orina refluye y drena a vejiga sin dificultad (véase sección RVU).
4. No obstruido, no refluyente. En estos pacientes existe una dilatación ureteral pero, paradójicamente, no presentan reflujo ni obstrucción(11).
Clínica. Representa el 23% de los casos de hidronefrosis en el RN de causa obstructiva. Es más frecuente en niños y más común en el lado izquierdo. Ocurre de manera bilateral en el 25%. Son diagnosticados incidentalmente en la ecografía prenatal y típicamente la exploración física es normal. Los casos que no son diagnosticados de forma prenatal suelen presentar posteriormente ITUs, hematuria, dolor abdominal, masa o uremia. Es raro que evolucione a IRT.
Diagnóstico. La ecografía es la primera prueba a realizar, confirma la dilatación ureteral. Se debe determinar mediante la realización de un MAG-3 qué megauréteres son obstructivos y, mediante una CUMS, cuáles tienen RVU, así como los casos en los que se combinen obstrucción y RVU. Incluso los pacientes que no tienen RVU ni obstrucción requieren ser identificados para un seguimiento estrecho(12).
Tratamiento. No hay parámetros que determinen qué pacientes van a requerir cirugía. Dado que el 70% de los casos se resuelven espontáneamente antes de los 2 años y que las intervenciones quirúrgicas tempranas se relacionan con tasa de complicación altas, no se recomienda el tratamiento quirúrgico excepto en megauréteres con ITUs recurrentes, deterioro de la FRD u obstrucción significativa. La edad idónea para realizar la reparación quirúrgica será entre el año de vida y los 2 años(13,14).
Seguimiento. Si permanece estable, se debe seguir de 2 a 4 años con ecografía y MAG-3 cada 6-12 meses(15). Tras la intervención, se debe solicitar una CUMS y MAG-3 más allá de los 3 meses para descartar RVU y obstrucción, respectivamente.
Duplicidad renal: ureterocele y uréter ectópico
La duplicación del sistema renal es una de las anomalías congénitas más frecuentes del tracto urinario, cursando de forma asintomática.
Embriología: las duplicaciones del tracto urinario superior son el resultado del desdoblamiento de la yema ureteral primitiva. De cada conducto de Wolf surge una yema ureteral que se une con el blastema metanéfrico para formar el riñón, y al seno urogenital para formar la vejiga. El grado de duplicación ureteral depende del momento en el que tiene lugar la separación inicial de las yemas; mientras que, el de la duplicación renal tiene relación con la distancia que existe entre ellas antes de alcanzar el blastema metanéfrico. Si brotan varias yemas ureterales, la ley de Weigert-Meyer indica que el uréter que drena la porción superior del riñón (uréter del hemisistema superior), termina de manera más caudal y medial que el que drena su porción inferior (uréter del hemisistema inferior). El desplazamiento del uréter superior de su posición habitual hace más probable que desemboque ectópicamente y se asocie a un ureterocele que condicione obstrucción; mientras que, el menor tiempo de desarrollo del uréter inferior disminuye su capa muscular, facilitando la aparición de RVU.
Ureterocele
Es el resultado de la dilatación quística del segmento terminal intravesical del uréter (Fig. 2). La forma más frecuente de presentación es una ITU en los primeros meses de vida. La mayoría precisan tratamiento quirúrgico, siendo la punción endoscópica la técnica de elección.

Figura 2. Imagen del ureterocele.
Epidemiología. Es 4 a 7 veces más frecuente en mujeres de raza blanca, con un 10% de los casos bilaterales. Suele estar asociado al pielón superior de un doble sistema completo (80%).
Clínica. La forma más frecuente de presentación es una ITU en los primeros meses de vida. Otros son detectados incidentalmente en ecografía prenatal o como una masa abdominal palpable secundaria a obstrucción renal. Aunque la obstrucción uretral es rara, una de sus causas más frecuentes en niños es el prolapso uretral de un ureterocele.
Diagnóstico. La primera prueba a efectuar, la ecografía, muestra una masa quística intravesical bien definida en la parte posterior de la vejiga. Se puede ver también un uréter proximal dilatado y, muy a menudo, un riñón doble ipsilateral en el que el hemirriñón superior está displásico. La CUMS se debe realizar siempre, ya que, en más del 50% de los casos, el pielón inferior ipsilateral y en el 25% del contralateral tienen RVU. Además, se puede observar el ureterocele como un defecto de llenado en la vejiga en las primeras imágenes de la prueba. El DMSA permite valorar la función del hemirriñón superior y el MAG-3 la eliminación cuando se sospecha obstrucción. La UIV, en desuso, mostrará el “signo de la cobra” en la vejiga y el “signo de la flor marchita” si hay sistema doble.
Tratamiento. Éste depende del tipo de ureterocele y del modo de presentación. La mayoría de pacientes precisan cirugía. La primera opción terapéutica es la punción endoscópica, con un éxito del 90% en los ureteroceles intravesicales y 50% en los ectópicos y, si ésta falla, se debe realizar una reconstrucción por cirugía abierta. En caso de anulación funcional del pielón superior, el procedimiento de elección es la heminefrectomía, habitualmente por cirugía mínimamente invasiva(16). En los pacientes con sepsis secundaria a obstrucción, el tratamiento inmediato es la nefrostomía.
Uréter ectópico
Es el uréter cuyo meato termina en una posición caudal a la inserción normal del uréter en el trígono. Es más frecuente en mujeres, manifestándose como ITU e incontinencia en niñas mayores.
El orificio se encuentra siempre a lo largo del trayecto del desarrollo normal del sistema mesonéfrico, por tanto, en niños es en el cuello vesical, próstata (a nivel del orificio del conducto eyaculatorio) o, incluso, a lo largo del trayecto del sistema genital masculino, incluyendo el epidídimo. En niñas, el orificio puede terminar en el cuello vesical, uretra, vagina o, de forma más rara, en cérvix y útero.
Epidemiología. Más frecuente en mujeres, un 70% asocia duplicación ureteral completa con una alta incidencia de tejido renal displásico en el hemirriñón superior correspondiente al uréter ectópico(11).
Clínica. Lo más frecuente son ITUs de repetición. Dependiendo del sexo, observaremos incontinencia en niñas mayores, con pérdida de orina a pesar de tener un buen hábito miccional. Esto no ocurre en el varón; ya que, siempre desemboca antes del esfínter uretral externo. Si el uréter drena en el conducto genital, puede provocar orquiepididimitis. En ambos casos, se puede apreciar HN en la etapa prenatal si el orificio ureteral está obstruido, por ejemplo, secundario al drenaje a nivel del epidídimo en el varón o del esfínter en la mujer, o un riñón displásico si el uréter es extrauretral.
Diagnóstico. En la ecografía se aprecia un uréter distal dilatado. En los casos en los que no esté dilatado, por ejemplo en niñas mayores en las que el uréter drena a la vagina, el TC o RNM con contraste pueden determinar con precisión la localización del uréter. La cistoscopia normalmente no es diagnóstica por sí sola, pero puede ser útil para identificar el ostium ureteral en la uretra, lo cual es muy complejo si el orificio está dentro de la vagina.
Tratamiento. Diferenciamos dos situaciones en el tratamiento del uréter ectópico:
• Sistema doble: como la mayoría de los casos están asociados con un polo superior displásico; la exéresis de este segmento y del uréter proximal es normalmente curativa.
• Sistema único: en niñas, el riñón asociado con el uréter ectópico es normalmente pequeño y con función muy pobre. Si el riñón es funcionante, el tratamiento es la resección del uréter ectópico distal y la reimplantación. Si no, se realiza nefroureterectomía por cirugía abierta o laparoscopia.
Válvulas de la uretra posterior (VUP)
Las VUP son unos pliegues membranosos dentro de la luz uretral de los varones, siendo una de las causas más frecuentes de obstrucción del tracto urinario, pudiendo ocasionar en el periodo neonatal un amplio espectro de severidad, ocurriendo en 1/5.000-8.000 embarazos(17).
Fisiopatología. La obstrucción de salida al flujo urinario produce hipertrofia y depósito de colágeno en el detrusor que ocasionan alteración de la compliance o acomodación vesical, aumentando la presión intravesical que se transmite a todo el TUS, pudiendo ocasionar ureterohidronefrosis, RVU y lesión renal (disfunción glomerular y tubular) (Fig. 3). Sin embargo, si se alivia la obstrucción de forma temprana, estos cambios pueden revertirse, excepto cuando exista displasia renal acompañante(18). El RVU está presente entre 1/3 y 1/2 de los pacientes con VUP. El RVU unilateral actúa como mecanismo de escape de la presión intravesical, salvaguardando la función del riñón contralateral a corto plazo, denominándose síndrome VURD(19) (vesicoureteral reflux unilateral renal dysplasia), aunque con el tiempo no parece que la preserve. Otros mecanismos de descarga de presión del TUS incluyen grandes divertículos vesicales, ascitis urinaria prenatal debida a la extravasación calicial y urinoma penirrenal. La disfunción vesical se presenta, según las distintas series, entre un 15 a 70% de los casos con VUP. Las alteraciones vesicales presentes constituyen el denominado “síndrome de la vejiga valvular”, existiendo tres patrones urodinámicos: hiperactividad del detrusor, baja acomodación vesical y fallo miogénico(20). Para algunos autores, estos patrones representan diferentes etapas de un mismo proceso. El más frecuente es la hiperactividad del detrusor (70%), siendo los 2 últimos los de peor pronóstico, evolucionando un 50% al fracaso renal con la necesidad de trasplante.
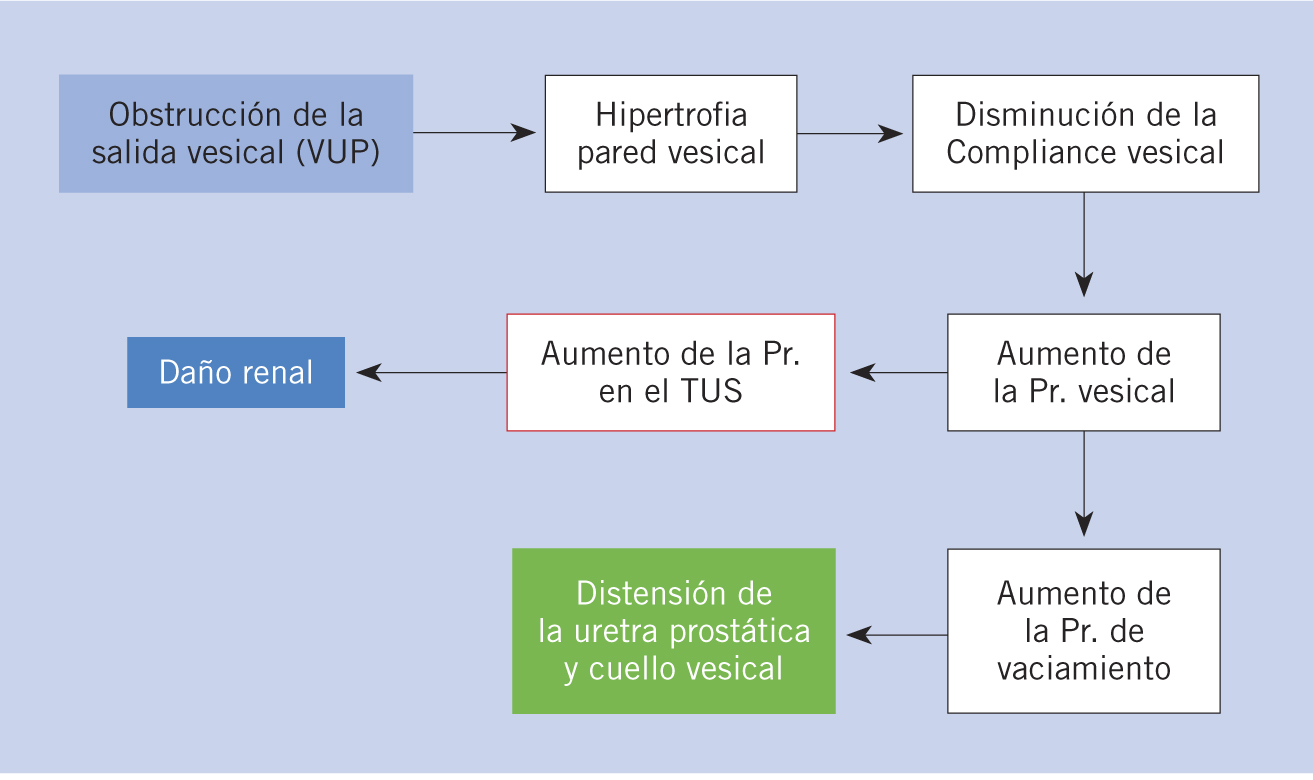
Figura 3. Fisiopatología de las válvulas de uretra posterior (VUP) (A. Gómez Fraile).
Diagnóstico. En la actualidad, la mayoría de las VUP se detectan por ecografía prenatal. Este descubrimiento permite el tratamiento temprano postnatal. Las imágenes sugestivas de VUP son la ureterohidronefrosis bilateral, aumento del tamaño de la vejiga y uretra posterior dilatada. En ocasiones, veremos engrosamiento de la pared vesical (>3 mm) y signos de displasia renal (aumento de la ecogenicidad renal y quistes corticales). La CUMS confirma el diagnóstico al nacimiento. La uretra posterior presenta una importante dilatación y el cuello de la vejiga está estrechado y engrosado con el canal urinario desplazado hacia la parte anterior (Fig. 4). El RVU suele ser intenso y aparece en la mitad de los lactantes. El DMSA y el MAG-3 valoran el estado del parénquima renal y el grado de obstrucción, respectivamente. Los estudios de laboratorio incluyen la determinación de urea, creatinina y electrolitos para valorar la función renal. Una creatinina mayor de 0,8-1 mg/dl, al año de la ablación valvular, sugiere deterioro de la TGF (<70 ml/min/1,73 m2) y función renal deficiente a largo plazo(21).
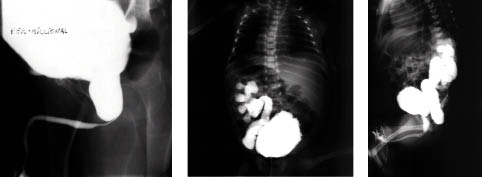
Figura 4. En la CUMS se aprecia gran dilatación en la uretra prostática, vejiga de lucha y gran reflujo vésico-ureteral unilateral.
Tratamiento prenatal. Cuando el oligohidramnios está presente en el 2º trimestre del embarazo, la mortalidad perinatal puede llegar al 90-95%. Por ello, la cirugía fetal puede considerarse en fetos con alto riesgo de muerte intraútero o neonatal debido a la existencia de un oligohidraminos severo, debiendo tener un cariotipo normal y una buena función renal estudiada en muestras de orina tomadas por punción de la vejiga fetal. En estos casos, la cirugía debería ser realizada en centros con gran experiencia debido al riesgo de mortalidad fetal (43%). Sin embargo, los resultados a largo plazo indican que la intervención prenatal no parece que mejore la función renal(22).
Tratamiento postnatal. Se inicia con la evaluación radiológica (ecografía, CUMS, MAG-3) y colocación de un catéter vesical para el drenaje del tracto urinario, pautando profilaxis antibiótica mientras se mantenga ésta y se descarte la presencia de RVU. Otras opciones de cirugía derivativa son la vesicostomía previa a la resección valvular, frecuentemente en niños prematuros, la pielostomía o ureterostomía alta, raramente realizadas debido a que precisan la reconstrucción posterior del TUS. Por otro lado, alteran el ciclo miccional de la vejiga, lo cual puede afectar a la función vesical, no mejorando los resultados de la vesicostomía. Podrían estar indicadas cuando, a pesar de un buen drenaje vesical, persiste la alteración de la función renal o dilatación ureteral. Una alternativa es la nefrostomía bilateral temporal. El tratamiento de elección, definitivo, se basa en la cistoscopia diagnóstica y terapéutica, mediante ablación endoscópica de las VUP, para la resolución de la obstrucción uretral(20).
Seguimiento. Después del procedimiento quirúrgico de las válvulas de uretra, el tratamiento incluye la detección de la disfunción vesical mediante pruebas de imagen y estudio urodinámico, además de la monitorización de la función renal. Un tercio de los pacientes con disfunción vesical severa requerirán sondaje limpio intermitente y medicación anticolinérgica y/o a-bloqueante, dependiendo de los resultados de la urodinamia. A pesar del diagnóstico prenatal y de la intervención precoz, un 15 a 20% progresan hacia la IRT, precisando trasplante renal(21).
Reflujo vésico-ureteral (RVU)
Proceso heterogéneo y multifactorial en el que se produce el paso retrógrado, no fisiológico, de orina al TUS. Anatómicamente, el uréter terminal atraviesa la pared vesical en un túnel submucoso que desemboca en el trígono. Mediante la presión intravesical, la porción terminal del uréter es comprimida contra el detrusor, creando un mecanismo valvular pasivo que impide el ascenso de orina.
Prevalencia. En población sana es desconocida. Se asume una prevalencia del 1% en RNV y muy superior, hasta un 30-70%, tras una ITU febril.
Grupos. Raza: más frecuente en niños de raza blanca presentando, además, mayor grado de RVU. Edad: mayor prevalencia cuanto menor es el niño. Sexo: después del primer año, la prevalencia de ITU es 2 veces superior en la mujer. Sin embargo, los niños tienen, entre todos los pacientes con ITU, más probabilidad de tener RVU (hombre:mujer de 2:1), además de ser normalmente diagnosticados durante el periodo prenatal. Los varones con antecedentes familiares de RVU tienen mayor riesgo de daño congénito(23) (Tabla II).

Genética. Existe un importante componente familiar, evidenciado por la alta incidencia de RVU en hermanos (30%) y en hijos de padres con RVU (35-50%). Sólo estaría indicado el estudio genético en caso de: hipoplasia, displasia, fallo renal crónico no explicado o alteraciones extrarrenales que puedan tener algún significado sindrómico.
Tipos de RVU. Tradicionalmente, el RVU se ha clasificado en primario y secundario:
• Primario: debido a un debilitamiento del funcionamiento del sistema valvular antirreflujo por inmadurez o alteración de la unión vésico-ureteral. Dentro de este grupo, podemos discernir 2 entidades: el reflujo congénito, diagnosticado por ecografía prenatal, y el RVU tras una ITU.
• Secundario: asociado a un funcionamiento anómalo de la vejiga con presiones intravesicales altas (vejiga neuropática o VUP). Este RVU tiende a mejorar cuando las presiones vesicales se normalizan.
Clínica. La ITU representa la forma más común de presentación (30%). Un síntoma asociado es el dolor, aunque se debe más a su relación con la pielonefritis. En alguna ocasión, el RVU se manifiesta como IRT y/o HTA en pacientes con poca o ninguna historia de ITU previa, lo cual podría ser secundario a una displasia renal congénita. Desde el advenimiento de la ecografía, debemos incluir las formas asintomáticas como otra opción de debut, así como los RVU prenatales.
Diagnóstico. Existen diferentes técnicas de imagen para diagnosticar y graduar el RVU: la cistografía isotópica directa, la ecocistografía con contraste y la CUMS. Sin embargo, es esta última la que, pese a su invasividad y radiación, continúa siendo el gold estándar, proporcionando un detalle anatómico superior, muy importante en varones, y valorando la presencia de reflujo intrarrenal. La clasificación más admitida, basada en la magnitud del paso retrógrado de orina de la vejiga al uréter, es la proporcionada por el International Reflux Study Committee(24). En general, se acepta que los RVU grado I y II son leves; el grado III moderado y los grados IV y V graves (Tabla III). Otra prueba complementaria es el DMSA, la más sensible para visualizar cicatrices y cuantificar la FRD(25). Para evaluar las cicatrices nos podemos guiar por la clasificación de Goldraich. Un RVU con un DMSA normal es de bajo grado y tiene un elevado porcentaje de resolución. Screening en hermanos: aunque sería interesante, éste no se suele llevar a cabo debido a la invasividad de las pruebas. La ecografía continúa siendo, pese a su baja sensibilidad (<50%), la prueba de elección.

Nefropatía por RVU (NR). Puede ocurrir en la vida fetal en forma de hipoplasia-displasia pero, en su mayoría, ocurre después del nacimiento y, sobre todo, en relación con la ITU. Es más frecuente en mujeres y se ha asociado a HTA, IRC, IRT y complicaciones en el embarazo.
Tratamiento. El enfoque terapéutico del RVU ha cambiado, adoptando una actitud más conservadora, como consecuencia de la falta de evidencia de beneficio significativo del tratamiento quirúrgico frente al médico. Diversos estudios han demostrado que la evolución natural del RVU es hacia la corrección espontánea, a razón de 10-15% por año. De este modo, el grado del RVU es el principal factor predictor a la hora de valorar la probabilidad de resolución espontánea. Además, se sabe que el RVU no suele dañar el riñón en ausencia de pielonefritis aguda asociada (PNA). El tratamiento conservador debe considerarse el modelo terapéutico de elección inicial (Fig. 5)(26).
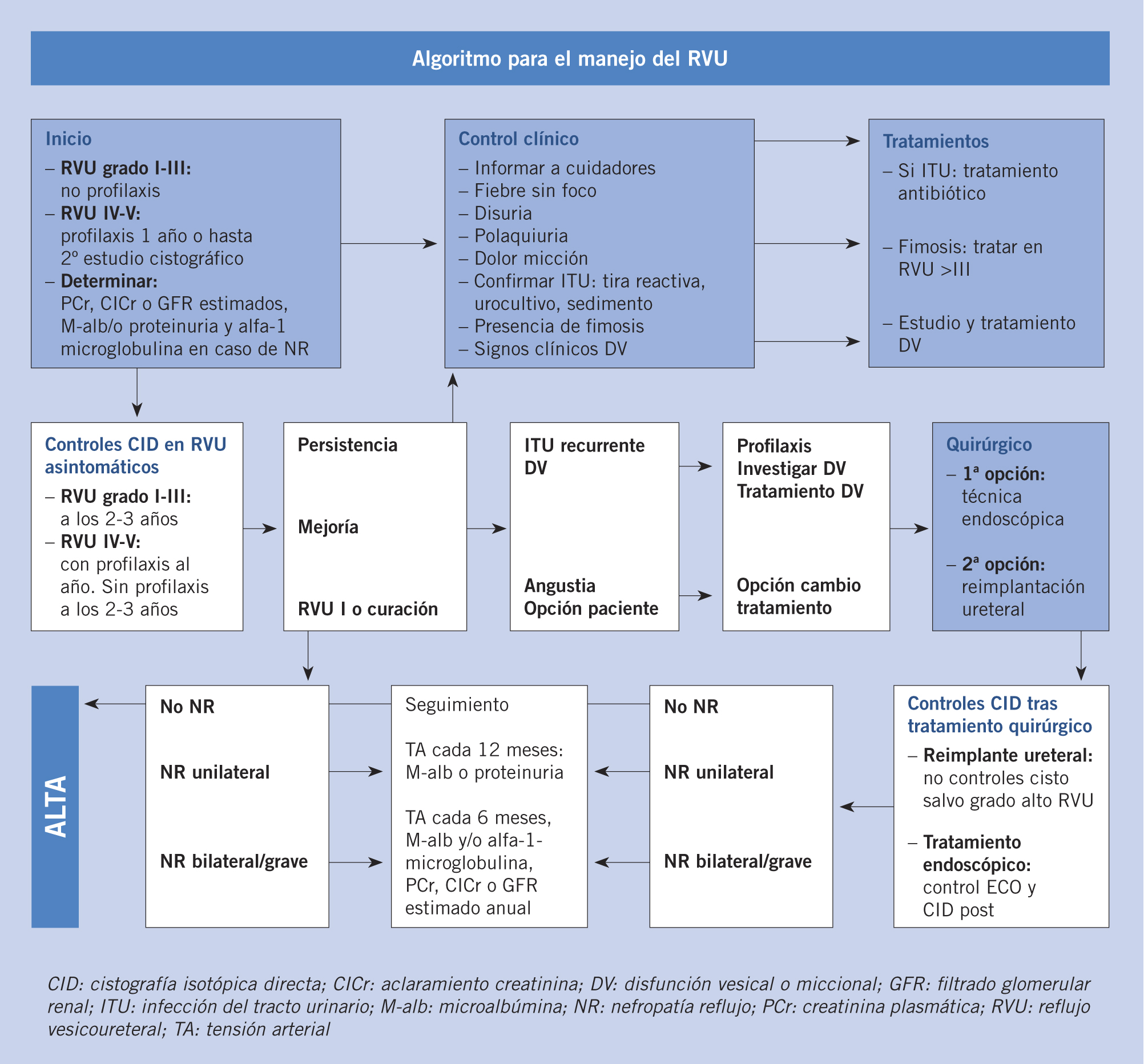
Figura 5. Manejo del reflujo vésico-ureteral (RVU).
• Médico:
– Medidas generales para evitar la proliferación de gérmenes: estimulación de hábitos miccionales correctos con micciones completas y frecuentes, así como ingesta de abundantes líquidos y corrección del estreñimiento.
– Profilaxis antibiótica: no se recomienda de forma sistemática, debido a que no disminuye la progresión de daño renal(27). Se debe administrar en niños con RVU primario grado I-III con ITU de repetición y en los grados IV-V durante 1 año o hasta efectuar la siguiente CUMS, siendo el trimetoprim a 1-2 m/kg/día en dosis única nocturna el más utilizado.
• Quirúrgico: a mayor grado de RVU, menor probabilidad de resolución espontánea con mayor riesgo de daño renal. El tratamiento quirúrgico ha demostrado ser útil en reducir el número de PNA, pese a que no disminuye la progresión del daño renal. Por tanto, la principal indicación quirúrgica, y que debe plantearnos cambiar la actitud terapéutica, es la PNA. Para ello, disponemos de 2 tipos de cirugías, la reimplantación ureteral, generalmente abierta, más invasiva, pero con porcentajes de éxito >95%, y el tratamiento endoscópico, menos invasivo y con un porcentaje variable de éxito en función del número de procedimientos realizados, edad y sexo del paciente, grado de RVU y anatomía vésico-ureteral, ya que los dobles sistemas renales son más resistentes a la curación(28). Con esta técnica, se crea un habón submucoso a nivel del ostium ureteral mediante la inyección de un material (Deflux®, Macroplastique®), que disminuye el orificio y dificulta el paso retrógrado de orina.
Bibliografía
Los asteriscos reflejan el interés del artículo a juicio del autor.
1. Queisser-Luft A, Stolz G, Wiesel A, Schlaefer K, Spranger J. Malformations in newborn: results based on 30,940 infants and fetuses from the Mainz congenital birth defect monitoring system (1990-1998). Arch Gynecol Obstet. 2002; 266(3): 163-7.
2. Onal B, Kogan BA. Natural history of patients with multicystic dysplastic kidney-what followup is needed? J Urol. 2006; 176(4 Pt 1): 1607-11.
3. Narchi H. Risk of Wilms’ tumour with multicystic kidney disease: a systematic review. Arch Dis Child. 2005; 90(2): 147-9.
4. Neville H, Ritchey ML, Shamberger RC, Haase G, Perlman S, Yoshioka T. The occurrence of Wilms tumor in horseshoe kidneys: a report from the National Wilms Tumor Study Group (NWTSG). J Pediatr Surg. 2002; 37(8): 1134-7.
5.*** Lupton EW, Testa HJ, O’Reilly PH, Gosling JA, Dixon JS, Lawson RS, et al. Diuresis renography and morphology in upper urinary tract obstruction. Br J Urol. 1979; 51(1): 10-4.
6. Fernbach SK, Maizels M, Conway JJ. Ultrasound grading of hydronephrosis: introduction to the system used by the Society for Fetal Urology. Pediatr Radiol. 1993; 23(6): 478-80. Disponible en: http://www.uab.edu/images/peduro/SFU/sfu_grading_on_web/sfu_grading_on_web.htm.
7.*** Essentials of paediatric urology. second ed. Informa Healthcare; 2008.
8.*** Parente HA, Angulo Madero JM, Romero Ruiz RM, Rivas VS, Lain FA, Fanjul GM. Medium-term results of the endourological management with balloon dilatation of the ureteropelvic junction stenosis in infants. Actas Urol Esp. 2009; 33(4): 422-8.
9. Kang HJ, Lee HY, Jin MH, Jeong HJ, Han SW. Decreased interstitial cells of Cajal-like cells, possible cause of congenital refluxing megaureters: Histopathologic differences in refluxing and obstructive megaureters. Urology. 2009; 74(2): 318-23.
10. Shokeir AA, Nijman RJ. Primary megaureter: current trends in diagnosis and treatment. BJU Int. 2000; 86(7): 861-8.
11. Michael DiSandro. Hydroureteronephrosis. En: Laurence S, Baskin BAK, editores. Handbook of pediatric urology. 2nd Edition ed. Lippincott Williams & Wilkins; 2005.
12. Hodges SJ, Werle D, McLorie G, Atala A. Megaureter. ScientificWorldJournal. 2010; 10: 603-12.
13. Arena F, Baldari S, Proietto F, Centorrino A, Scalfari G, Romeo G. Conservative treatment in primary neonatal megaureter. Eur J Pediatr Surg. 1998; 8(6): 347-51.
14. Peters CA, Mandell J, Lebowitz RL, Colodny AH, Bauer SB, Hendren WH, et al. Congenital obstructed megaureters in early infancy: diagnosis and treatment. J Urol. 1989; 142(2 Pt 2): 641-5.
15. Trobs RB, Heinecke K, Elouahidi T, Nounla J, Kluge R. Renal function and urine drainage after conservative or operative treatment of primary (obstructive) megaureter in infants and children. Int Urol Nephrol. 2006; 38(1): 141-7.
16. Nerli RB, Vernekar R, Guntaka AK, Patil SM, Jali SM, Hiremath MB. Laparoscopic hemi/partial nephrectomy in children with ureteral duplication anomalies. Pediatr Surg Int 2011; 27(7): 769-74.
17. Brown T, Mandell J, Lebowitz RL. Neonatal hydronephrosis in the era of sonography. AJR Am J Roentgenol. 1987; 148(5): 959-63.
18. Gómez Fraile A, López Vázquez F. Alteraciones del tracto de salida vesical. En: Gutiérrez Segura C, editor. Patología funcional urológica y urodinámica pediátrica. Siglo SL; 2008.
19. Hoover DL, Duckett JW, Jr. Posterior urethral valves, unilateral reflux and renal dysplasia: a syndrome. J Urol. 1982; 128(5): 994-7.
20.*** Casal AJ. Válvulas uretrales posteriores y otras anomalías uretrales. En: Campbell-Walsh, editor. Urology. Panamericana; 2007.
21. Ansari MS, Gulia A, Srivastava A, Kapoor R. Risk factors for progression to end-stage renal disease in children with posterior urethral valves. J Pediatr Urol. 2010; 6(3): 261-4.
22. Holmes N, Harrison MR, Baskin LS. Fetal surgery for posterior urethral valves: long-term postnatal outcomes. Pediatrics. 2001; 108(1): E7.
23. Essentials of pediatric radiology. A multimodality approach. 1ª ed. Cambridge; 2010.
24. Lebowitz RL, Olbing H, Parkkulainen KV, Smellie JM, Tamminen-Mobius TE. International system of radiographic grading of vesicoureteric reflux. International Reflux Study in Children. Pediatr Radiol. 1985; 15(2): 105-9.
25. Espino Hernández MM, Loris Pablo C. Reflujo vesicoureteral primario. Protocolos diagnósticos terapéuticos de la AEP 2008. Nefrología.
26. Areses Trapote R, Escribano Subías J, Fraga Rodríguez GM, Gracia Romero J, Loris Pablo C, Valenciano Fuente B. GPC Reflujo vésico-ureteral. Asociación Española de Nefrología Pediátrica; 2009 (GuíaSalud). Disponible en: http://www.guiasalud.es/egpc/reflujo/completa/apartado01/introduccion.html
27. Mori R, Fitzgerald A, Williams C, Tullus K, Verrier-Jones K, Lakhanpaul M. Antibiotic prophylaxis for children at risk of developing urinary tract infection: a systematic review. Acta Paediatr. 2009; 98(11): 1781-6.
28.*** Boston children Hospital 2013Available from: URL: http://www.childrenshospital.
org/vurcalculator/.
Bibliografía recomendada
– Lupton EW, Testa HJ, O’Reilly PH, Gosling JA, Dixon JS, Lawson RS, et al. Diuresis renography and morphology in upper urinary tract obstruction. Br J Urol. 1979; 51(1): 10-4.
Artículo original que describe las curvas de eliminación del renograma diurético descritas por O´Reilly.
– Essentials of paediatric urology. second ed. Informa Healthcare; 2008.
Libro clásico de la urología pediátrica que expone de manera sencilla y didáctica sus principales patologías.
– Parente HA, Angulo Madero JM, Romero Ruiz RM, Rivas VS, Lain FA, Fanjul GM. Medium-term results of the endourological management with balloon dilatation of the ureteropelvic junction stenosis in infants. Actas Urol Esp. 2009; 33(4): 422-8.
Este artículo refleja la la experiencia del Hospital Gregorio Marañón en el tratamiento endourológico de la estenosis pielo-ureteral.
– Casal AJ. Válvulas uretrales posteriores y otras anomalías uretrales. En: Campbell-Walsh, editor. Urology. Panamericana; 2007.
Compendio de urología, referencia en su campo, traducido al castellano. Cuenta con un tomo dedicado exclusivamente a la urología pediátrica.
– Boston children Hospital 2013. Available from: URL: http://www.childrenshospital.
org/vurcalculator/.
Página del Boston´s Children Hospital que permite la estimación de la efectividad del tratamiento endoscópico tras la cumplimentación de una serie de parámetros.
| Caso clínico | |
|
Presentamos el caso clínico de un RN varón con diagnóstico prenatal de hidronefrosis bilateral en la semana 20 de gestación, con un DAP máximo de 10 mm que en ecografías sucesivas ha ido paulatinamente disminuyendo, sin otros hallazgos ecográficos de interés. No constan antecedentes personales ni familiares reseñables. Nace por parto espontáneo, vaginal, en la semana 38 de gestación, con un Apgar 8/9 y un peso de 3.450 g. En la ecografía postnatal, se aprecia un riñón derecho de 54 mm, con buena diferenciación córtico-medular y un riñón izquierdo de 64 mm, con una cortical y diferenciación córtico-medular preservadas, con ureterohidronefrosis izquierda. La pelvis renal mide 15 mm, presentando un uréter mínimamente prominente a nivel distal (3 mm). Con el fin de descartar RVU, se solicita una CUMS, en la que se observa una vejiga de paredes finas sin defectos de repleción, sin RVU y con una uretra de características normales. El renograma diurético MAG-3 descarta obstrucción y muestra una FRD mantenida, lo que nos permite seguir al paciente en consultas. Durante este tiempo, el paciente se mantiene clínicamente asintomático. Las ecografías muestran en alguna ocasión un aumento de la dilatación piélica, pero que no se correlaciona con cambios en el MAG-3, los cuales mantienen un patrón no obstructivo, desapareciendo la hidronefrosis en las siguientes ecografías. Por otra parte, la dilatación ureteral ha ido disminuyendo hasta no ser perceptible. A los 4 años de edad, acude a revisión, aportando una ecografía en la que se aprecia una importante hidronefrosis izquierda que ha aumentado con respecto al estudio previo, midiendo la pelvis renal 46 milímetros de DAP. Así mismo, en la ecografía Doppler se visualizan unos vasos de gran calibre cruzando la pelvis renal. Ante estos hallazgos se decide solicitar un renograma diurético, en el que se observa una FRD del riñón izquierdo del 47% sin prácticamente respuesta a la administración del diurético, por lo que se interviene al paciente con el diagnóstico de estenosis pieloureteral secundaria a vasos polares.

Figura 6. Resultados del renograma diurético administrando furosemida en el minuto 20.
|
|

