Enfermedades por alteración de la impronta genética. Síndrome de Prader Willi y de Angelman
Introducción
Estudiamos de forma conjunta el SPW y el SA, al compartir la misma región cromosómica 15q11q13. Fueron los primeros síndromes relacionados con alteraciones de la impronta genómica con diferentes mecanismos genéticos que conducen a su presentación.
Las manifestaciones clínicas en el SPW, se deben a la falta de la información genética contenida en la región 15q11q13 que deriva del padre. La mayoría de los pacientes presentan una deleción de la región 15q11q13 de origen paterno (75-80%), otros una disomía uniparental materna, UPD(15)mat, de manera que los dos cromosomas 15 proceden de la madre y ninguno del padre (20-25%); por último, la causa puede residir en un defecto de la impronta (1-3%), el paciente tiene un cromosoma de cada progenitor, pero se ha establecido una impronta genómica incorrecta, el cromosoma paterno lleva una impronta materna, silenciando los genes de expresión paterna.
El fenotipo en el SA, se debe a la falta de la información genética materna de la región 15q11q13. La causa más frecuente es la deleción materna (70-75%). La mutación en la copia materna del gen UBE3A (10%), segunda causa por frecuencia. La disomía uniparental paterna UPD(15)pat es poco frecuente (3-7%), asi como los defectos de la impronta (2-4%); en este último caso, el cromosoma materno lleva una impronta paterna, silenciando los genes de expresión materna.
El diagnóstico de las dos entidades es clínico y debe ser confirmado mediante estudios moleculares. Es importante un diagnóstico precoz, para establecer un manejo específico del paciente. Establecer qué mecanismo genético es el responsable de estos síndromes permite ofrecer un correcto asesoramiento genético(1-5).
La prevalencia del SPW se sitúa entre 1:10.000-1:30.000 recién nacidos, la mayoría de los casos de presentación esporádica(4). La prevalencia para el SA se sitúa entre 1:15.000-1:20.000, probablemente está infradiagnosticado(6); en nuestra práctica, diagnosticamos adultos con SA, en los que no se había identificado previamente el síndrome. En cambio, en edad pediátrica, la sensibilidad de los neuropediatras es muy alta, el último diagnóstico en nuestro centro corresponde a un lactante de 10 meses.
Los SPW y SA forman parte del grupo de las enfermedades raras, definidas por su baja prevalencia, <5 casos por 10.000 habitantes, la mayoría debutan en la edad infantil, los pediatras tienen un papel protagonista en su identificación y atención(7).
Síndrome de Prader Willi
Las manifestaciones clínicas son muchas, variadas y cambiantes con la edad. La alteración genética produce una disfunción hipotalámica responsable de la clínica.
Los bebes con SPW nacen más pequeños, 15-20% menos que sus hermanos(8). La hipotonía prenatal disminuye los movimientos fetales, aumenta los partos por cesárea por presentación podálica y falta del trabajo fetal. La hipotonía dificulta la lactancia en los primeros meses, siendo a veces necesaria la alimentación mediante sonda nasogástrica.
El llanto es débil y presentan letargia. La hipotonía mejora con la edad, pero persiste en forma de menor tono y masa muscular en los adultos(4).
La escoliosis se observa en un 30% de pacientes, antes de los 10 años(8).
Las características faciales incluyen: estrechez bifrontal, ojos almendrados, labio superior fino, comisuras bucales hacia abajo, manos y pies pequeños, borde ulnar recto. Hipoplasia genital con criptorquidismo en varones(9).
En la primera infancia, hay un retraso del desarrollo motor y del lenguaje. Trastornos del aprendizaje en grado variable o discapacidad intelectual se observan en la edad escolar(10).
La conducta es muy característica ya en los niños, con: rabietas, terquedad, conducta manipuladora y dificultad para cambiar de rutinas. Los adolescentes son caprichosos, manipuladores y obstinados. En los adultos, persisten las alteraciones conductuales y presentan alto nivel de frustración. El trastorno mental más prevalente en adultos con SPW, es el Trastorno Obsesivo Compulsivo (TOC)(4).
La disfunción hipotalámica da lugar al complejo trastorno endocrinológico presente en el SPW, con: déficit de la hormona de crecimiento, hipogonadismo, hipotiroidismo, insuficiencia adrenal y baja densidad ósea. El especialista en endocrinología pediátrico y luego el de adultos, se convierten en el eje vertebrador del manejo multidisciplinar(11).
La obesidad sigue siendo la mayor causa de morbilidad y mortalidad en el SPW, la obesidad se produce por la combinación de: hiperfagia, falta de sensación de saciedad, metabolismo bajo y baja actividad física(4). El diagnóstico precoz permite una intervención conductual y nutricional antes de que se inicie la hiperfagia, con buenos resultados en edad pediátrica.
Para identificar aquellos pacientes que podían tener SPW, se estableció un consenso de criterios clínicos para SPW (Holm, et al, 1993), pero quedaban un 20% sin diagnóstico. En la actualidad, se sigue el publicado por Gunay-Aygun et al, 2001 (Tabla I)(10).

El diagnóstico diferencial es amplio, pero si se siguen las indicaciones de la tabla I y como la prueba genética identifica el 100% de los casos, no deberíamos, en edad pediátrica, tener casos sin diagnóstico.
El diagnóstico precoz, el tratamiento con hormona de crecimiento y el manejo multidisciplinar ha mejorado considerablemente el pronóstico de las personas con SPW, pero nuevos retos se imponen para seguir cambiando la historia natural de la enfermedad(12).
Síndrome de Angelman
Las personas con SA presentan una discapacidad intelectual severa, con: grave afectación del lenguaje oral, dificultades motoras, trastorno del sueño, epilepsia y un fenotipo conductual específico(13).
Al nacer, no presentan dismorfias, pero al final del primer año, puede verse un estancamiento del perímetro craneal con: braquicefalia, boca grande con tendencia a la protrusión lingual, dientes pequeños y separados. Hipopigmentación en relación a su familia, en aquellos con deleción(5).
El diagnóstico se basa en los aspectos clínicos antes mencionados, aunque es muy superponible a otras entidades que afectan el neurodesarrollo. Los principales criterios clínicos están descritos en la tabla II.
En todos los pacientes, se observa retraso del desarrollo psicomotor en el primer año de la vida, entre los 2-5 años es evidente que es grave, con baja capacidad de atención e hiperactividad, necesitando cuidado y supervisión directa toda la vida(5,14).
La mayoría de casos con SA no adquirirán lenguaje oral o muy pocas palabras (unas 6), comprenden más de lo que pueden expresar, por ello pueden mejorar la comunicación con Sistemas de Comunicación Aumentativos y Alternativos (sistemas de CAA)(14).
Los niños empiezan a andar entre los 3-4 años, la marcha es muy característica, tendencia a caminar de puntillas, persiguiendo su centro de gravedad, especialmente evidente al correr. Los que tienen mayor espasticidad amplían la base de sustentación y se ayudan con los brazos levantados con flexión de los codos que recuerda un candelabro.
Son muy frecuentes los trastornos del sueño, afectan a un 40-80% de los individuos, en la conciliación y en el mantenimiento del mismo(5).
Más del 80% de los pacientes van a presentar epilepsia, con un patrón de EEG característico. La mayoría de los pacientes debutan antes de los 3 años, todo tipo de crisis han estado descritas, pero las más típicas son: las ausencias atípicas, la epilepsia mioclónica y el estatus epiléptico no convulsivo(6).
El fenotipo conductual es muy característico por: aspecto feliz, episodios de risa, atracción por el agua, llevarse objetos a la boca, conducta hiperactiva con falta de atención y conducta fácilmente excitable(14).
Es frecuente: el estreñimiento, la escoliosis y la obesidad en niños mayores y adultos. Atención con los problemas digestivos, como el reflujo gastroesofágico a cualquier edad(5).
El diagnóstico se basa en los criterios clínicos (Tabla II), aunque es superponible a otras muchas entidades que afectan al neurodesarrollo con importante afectación del lenguaje. Debe confirmarse con el test genético(6).

En un 10% de los pacientes con clínica de SA, el diagnóstico no se confirma molecularmente; en los últimos años, el diagnóstico diferencial (DD) se ha ido ampliando, incluyendo síndromes como: síndrome de Mowat-Wilson, síndrome de Pitt-Hopkins, síndrome de Phelan-McDermid y síndrome de Rett, entre otros. Nuestro equipo ha estudiado 16 pacientes con fenotipo SA-like en los que se había excluido los síndromes ya conocidos de DD, identificando en 12 de los 16, mutaciones en genes como: KIF1A, SYNGAP1, VAMP2… Un paciente presentaba una mutación en SMARCE1 asociado al síndrome de Coffin-Siris, por lo que proponemos que también se considere en el DD del SA(15).
Nuestra opinión es que este 10%, corresponde a otras entidades que se van identificando con las nuevas tecnologías de secuenciación masiva que amplían el diagnóstico diferencial, pero que no corresponden a SA.
El tratamiento es sintomático, incluye: manejo de la afectación motora, estrategias para mejorar la comunicación y manejo de la epilepsia y de las comorbilidades que puedan aparecer(16).
La esperanza de vida no se ve afectada y los síntomas son menos graves en el adulto(13).
Estructura y organización de la región cromosómica 15q11.2-q13
La región cromosómica 15q11.2-q13 ocupa aproximadamente una longitud de 6 Mb y contiene un clúster de genes que se encuentran regulados por el mecanismo de la impronta genómica. La impronta genómica es una marca epigenética que inactiva determinados genes en función de su origen parental. La inactivación o silenciamiento de los genes se produce por la metilación del ADN y el empaquetamiento de la cromatina, establecido por modificaciones de las histonas.
La ausencia de expresión de genes en el alelo paterno causa el SPW, mientras que la ausencia de un único gen, UBE3A, en el alelo materno causa el SA. La región 15q11.2-q13 está flanqueada por repeticiones de bajo número de copias que pueden originar deleciones mediante puntos de rotura (Break Points, BP) y recombinación homóloga desigual. Se conocen 3 puntos de rotura principales BP1 y BP2, que se encuentran más próximos al centrómero y BP3 que es más distal.
La región 15q11.2-q13 (Fig. 1) se puede subdividir en:

Figura 1. Región cromosómica 15q11.2-q13 y expresión diferencial de los genes de esta región según el origen parental. A. Genes expresados en el cromosoma paterno. B. Genes expresados en el cromosoma materno. Los recuadros en azul, indican genes de expresión paterna; los recuadros en rojo, indican genes de expresión materna; y los recuadros en gris, indican genes que se expresan de manera bialélica. Las flechas señalan la orientación de la transcripción. Los recuadros en blanco enmarcados en rojo o en azul, indician genes que no se expresan en el cromosoma paterno o materno, respectivamente. Las líneas en color azul, son los snoRNAs que están contenidos en el transcrito SNURF-SNRPN; y las líneas en color negro, indican los puntos de rotura BP. La región del centro de impronta (IC) está representada como cuadros enmarcados en rojo, el AS-SRO y en azul, el PWS-SRO. Los círculos en negro indican la metilación del ADN.
• Una región proximal sin impronta que se encuentra entre los puntos de rotura BP1 y BP2 y que contiene los genes NIPA1, NIPA2, CYFP1 y TUBGCP5, que se expresan de manera biparental(17).
• La región SPW, que solamente se expresa en el alelo paterno, incluye los genes: MKRN3, MAGEL2, NDN, C15orf2 y SNURF-SNRPN, este último contiene un clúster de genes small nucleolar RNA (SNORD107, SNORD64, SNORD108, SNORD109A, SNORD116, SNORD115 y SNORD109B) y varios transcritos antisentidos (incluyendo el transcrito antisentido del gen UBE3A, UBE3A-ATS). El promotor de estos genes se encuentra desmetilado en el cromosoma paterno en tejido cerebral. SNORD116 es clave para el desarrollo del SPW, pero también los genes: MAGEL2, con expresión en el hipotálamo, funciones en el ritmo circadiano, en el desarrollo de la estructura cerebral y en la reproducción e infertilidad; MKRN3, implicado en la regulación hormonal y en la pubertad precoz; y NDN, con funciones en el crecimiento axonal.
• La región SA que contiene los genes con expresión materna, UBE3A y ATP10A. UBE3A es responsable del SA.
• Una región distal entre BP2 y BP3 que no se encuentra sometida a impronta genómica, los genes incluidos en esta, se expresan de manera biparental. En esta región, se encuentra un clúster de genes receptores de GABA (GABRB3, GABRA5, GABRG3) y los genes OCA2 y HERC2.
La expresión génica en la región 15q11.2-q13 está regulada por un centro de impronta que se encuentra dividido en dos regiones críticas, el centro de impronta del síndrome de Prader Willi (PWS-SRO), que comprende el exón 1 del gen SNURF-SNRPN y una isla CpG que está sujeta a metilación diferencial. Se trata de una región comúnmente delecionada en familias SPW: en el alelo paterno, en los individuos afectados y, en el alelo materno, en el padre fenotípicamente normal. El centro PWS-SRO promueve la expresión de los genes del cromosoma paterno. El otro elemento es el centro de impronta del síndrome de Angelman (AS-SRO), que actúa reprimiendo el centro PWS-SRO en el cromosoma materno y se encuentra delecionado en familias SA, en el alelo materno en los individuos afectados y en el alelo paterno en las madres fenotípicamente normales(18,19).
Mecanismos moleculares y consejo genético
Se han descrito diferentes mecanismos moleculares que afectan a la región 15q11.2-q13 y que dan lugar a SPW y SA (Fig. 2):

Figura 2. Mecanismos moleculares del síndrome de Angelman (SA) y del síndrome de Prader-Willi (SPW). A. Mecanismos moleculares que originan el SPW y frecuencia de cada uno de ellos. B. Mecanismos moleculares causantes del SA y frecuencia de cada uno de ellos. Los cromosomas enmarcados en un fondo rojo, indican que son de origen materno o tienen un epigenotipo materno; y los cromosomas enmarcados en un fondo azul, indican un origen paterno o tienen un epigenotipo paterno. El asterisco en rojo, indica que se ha producido una deleción. La cruz en rojo, indica que no se expresa el gen UBE3A en el cromosoma materno. Mat: Materno; Pat: Paterno.
• Deleción de la región 15q11.2-q13: es la causa más frecuente, la deleción del alelo paterno representa el 75-80% de los casos de SPW y la deleción del alelo materno representa el 70-75% de los casos de SA. Se pueden distinguir principalmente dos tipos de deleciones: a) deleción tipo I, se encuentra en el 60% de los casos en SPW y en el 40% de los casos en SA y comprende desde el punto de rotura BP1 a BP3 (6 Mb); y b) deleción tipo II, que es el 30% de los casos en SPW y el 50% de los casos en SA, comprende desde el punto de rotura BP2 a BP3 (4 Mb)(20). Solo un 10% de las deleciones comprenden desde los puntos de rotura BP1/BP2 hasta puntos de rotura más distales como: BP4, BP5 o BP6(21,22). El riesgo de recurrencia es inferior al 1%.
• Disomía uniparental (UPD) del cromosoma 15: es la segunda causa más frecuente en el SPW (20-25%) y representa un 3-7% de los casos de SA. En el SPW, mayoritariamente se produce por una no disyunción meiótica materna, que da lugar a un ovocito disómico para el cromosoma 15 y después de la fertilización tendría lugar una pérdida mitótica del cromosoma paterno, ya que la trisomía del cromosoma 15 es letal. Este mecanismo se conoce como rescate trisómico y daría lugar a una heterodisomía(23). En el caso del SA, mayoritariamente son isodisomías, el origen de las cuales probablemente es debido a una no disyunción materna que resulta en una duplicación post-zigótica del cromosoma 15 paterno(24). De forma menos frecuente, se dan las isodisomías en SPW, que estarían producidas por una nulisomía del cromosoma 15, debido a una no disyunción meiótica, seguida de una duplicación post-zigótica del cromosoma 15 materno(25,26). El riesgo de recurrencia es inferior al 1%.
• Defecto de la impronta de la región 15q11.2-q13: es el mecanismo menos frecuente, representa un 1-3% de los casos de SPW y un 2-4% de los casos de SA. En la mayoría de los casos (85-90%), se produce por un defecto epigenético esporádico durante el establecimiento de la impronta en la gametogénesis o en el mantenimiento de la impronta después de la fertilización. El riesgo de recurrencia del defecto de impronta esporádico es inferior al 1%(27,28). En el 10-15% de los casos, el defecto de la impronta se produce por una deleción del centro de impronta SPW-SRO o AS-SRO, la mayoría de estos casos son familiares y tienen un riesgo de recurrencia del 50%(29).
• Mutaciones en el gen UBE3A: es la segunda causa más frecuente de SA (10%) y pueden ser de novo o familiares. Deleciones parciales o totales del gen UBE3A se han descrito también como causantes de SA, con una frecuencia inferior al 5% del total de mutaciones en UBE3A(30). En el caso de que sean familiares, el riesgo de recurrencia es del 50%.
En un 10% de los casos con diagnóstico clínico de SA, se desconoce la causa molecular y se recomienda realizar la secuenciación del exoma, ya que hay síndromes que presentan características clínicas solapantes con SA, como: síndrome de Kleefstra, síndrome de Rett, síndrome de Pitt Hopkins(31) o síndrome de Coffin-Siris, entre otros(15).
Estudios moleculares
El conocimiento de la causa genética del SPW y SA es imprescindible para poder orientar el pronóstico y el riesgo de recurrencia según el mecanismo molecular, y así poder ofrecer un asesoramiento genético. Además, en el caso del SPW, también es esencial un diagnóstico molecular, el más precoz posible, para iniciar el tratamiento con la hormona de crecimiento.
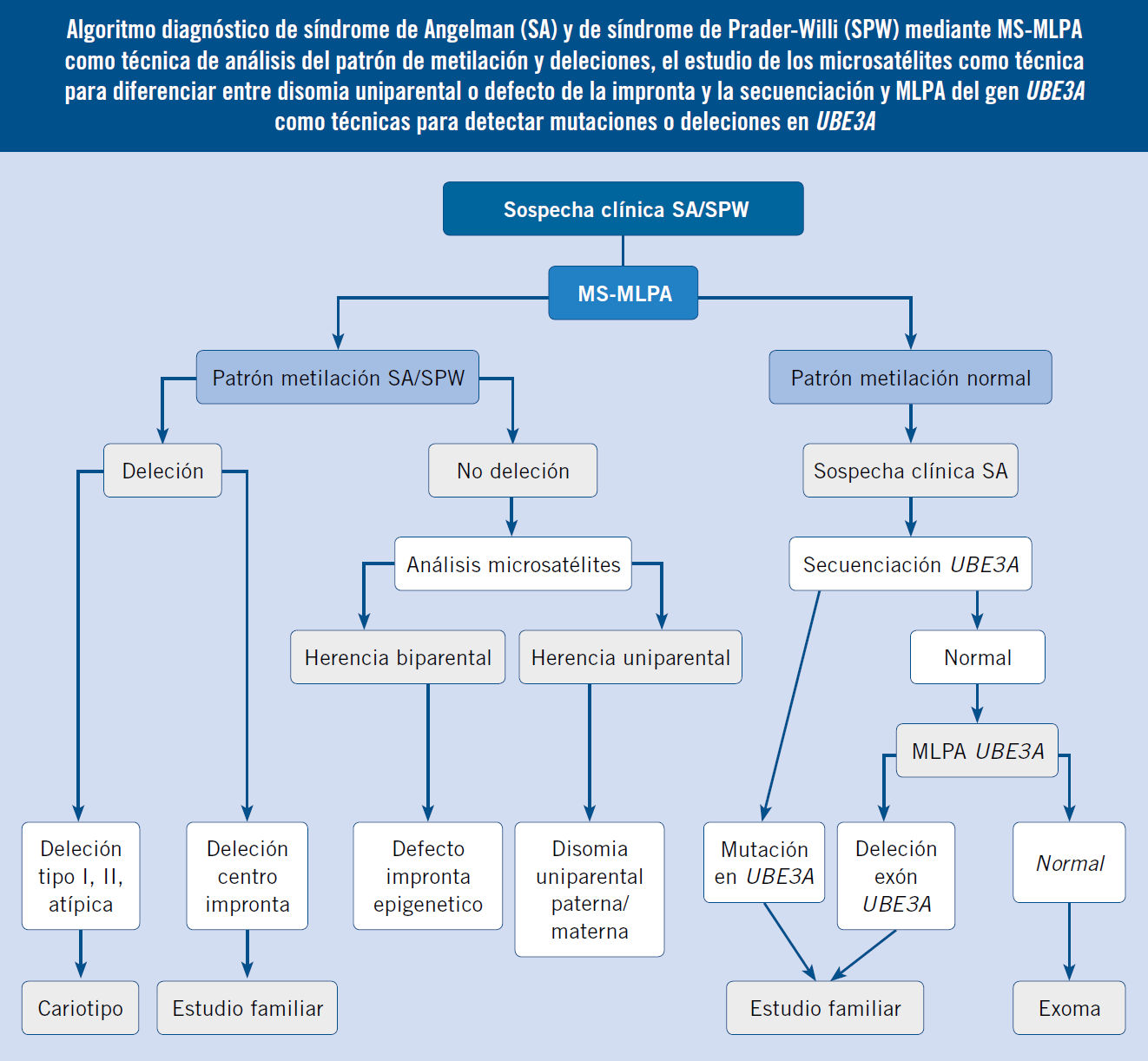
Frente a una sospecha clínica de SPW y SA, y teniendo en cuenta la frecuencia de las alteraciones moleculares, se recomienda iniciar el estudio analizando la metilación del centro de impronta (IC) de la región cromosómica 15q11.2-q13 (v. Algoritmo al final del artículo)(32).
• MS-MLPA (Methylation Specific-Multiplex Ligation-dependent Probe Amplification, MRC Holland). Esta metodología permite detectar cambios en el número de copias (CNVs) y la metilación del ADN en la región 15q11.2-q13. La última versión ME028-C1 contiene: 47 sondas, 34 de ellas dentro de la región 15q11.2-q13 (TUBGCP5, NIPA1, MKRN3, MAGEL2, NDN, SNRPN, UBE3A, ATP10A GABRB3)o cerca de la región crítica (APBA2 y OCA2). Como control, se utilizan 11 sondas situadas fuera de la región crítica de SPW y SA. Ocho sondas son sensibles a metilación y contienen un lugar de restricción de la enzima HhaI, seis se encuentran dentro de la región 15q11.2-q13 y dos se encuentran fuera de esta región y sirven como control de digestión. En el exón1- región promotora del gen SNURF-SNRPN, se encuentra una isla CpG que está metilada en el cromosoma materno y no metilada en el cromosoma paterno (Fig. 1), permitiendo valorar el patrón de metilación. En un individuo sano, el patrón de metilación es del 50%, se encuentra un alelo metilado (materno) y un alelo no metilado (paterno). En un individuo con SPW presenta un patrón de metilación del 100%, por: una deleción en el alelo paterno, una deleción en el centro de impronta (PWS-SRO), o bien, porque el alelo paterno tiene un epigenotipo materno. En un individuo con SA, el patrón de metilación es del 0%, ya que: se encuentra delecionado el alelo materno, hay una deleción en AS-SRO, o bien, el alelo materno tiene un epigenotipo paterno.
• Estudio de microsatélites del cromosoma 15. En los casos en los que haya un patrón de metilación de SPW o SA, pero no haya deleción en la región 15q11.2-q13, se requiere el análisis de microsatélites de esta región para distinguir si se trata de una disomía uniparental o un defecto de la impronta esporádico. Hay diferentes marcadores polimórficos dentro de esta región, que son adecuados para este propósito(33,34). Los microarrays de alta densidad con polimorfismos de un nucleótido (SNPs) también permiten proporcionar información sobre si hay una disomía uniparental. En el caso de que la herencia de los cromosomas 15 sea biparental, se concluye que el SPW o SA está producido por un defecto de la impronta esporádico.
En los casos que haya una sospecha clínica consistente de SA y el patrón de metilación sea normal (50%), se recomiendan realizar los siguientes estudios:
• Secuenciación del gen UBE3A. La secuenciación se puede llevar a cabo usando oligonucleótidos específicos que permitan amplificar cada uno de los exones del gen UBE3A, o bien, se puede secuenciar con la tecnología de secuenciación masiva (NGS) a través de paneles de genes dirigidos.
• MLPA del gen UBE3A. En el caso de que no se haya encontrado una mutación en el gen UBE3A que permita confirmar el diagnóstico, se recomienda realizar MLPA del gen UBE3A (P336-B1, MRC Holland). Esta técnica permite identificar deleciones parciales de exones o deleciones totales del gen UBE3A. El kit P336-B1 contiene: 37 sondas, una o dos para cada uno de los 10 exones de UBE3A; 3 sondas en el gen GABRB3, situado dentro de la región 15q11.2q13; y 4 sondas en el gen MTHFR, situado en la región 1p36.22.
Por último, en los casos en los cuales no se identifique ninguna de las causas genéticas conocidas y el paciente tenga un diagnóstico clínico de SA, estaría indicado realizar la secuenciación del exoma y estudiar los genes asociados a fenotipos similares, además de analizar el cariotipo molecular (array-CGH).
Correlación genotipo-fenotipo SPW
Los pacientes con deleción son los que presentan el fenotipo más grave, ya que se ha perdido un gran fragmento de ADN donde, además de los genes asociados con el SPW y regulados por impronta genómica, se encuentran otros genes(4,35,36). Los pacientes con deleción tipo I presentan un peor comportamiento adaptativo, mayor impulsividad, menor capacidad intelectual y rendimiento académico que los pacientes con deleción de tipo II(37). La conducta de: rascarse la piel, agresión, hiperfagia, así como un umbral alto para el dolor y alteraciones articulares, son más frecuentes y severas en los pacientes con deleción(38). La hipopigmentación está asociada a los casos con deleción por la pérdida del gen OCA2.
En los pacientes con UPD(15)mat, a diferencia de los pacientes con deleción, no ha ocurrido la pérdida física de ADN, sino la pérdida funcional de genes regulados por impronta genómica que se encuentran silenciados por metilación. Los pacientes con UPD tienen menos probabilidades de tener el aspecto facial típico, la hipopigmentación o la habilidad para realizar puzles(37). Presentan un mayor coeficiente intelectual verbal y mayor habilidad para el cálculo numérico. Los individuos con UPD tienen más probabilidades de desarrollar trastornos psiquiátricos, como psicosis afectiva y trastornos del espectro autista en edad adolescente-adulta, mientras que en la deleción, es más frecuente la depresión(39,40). Una disminución de materia gris en los ganglios basales, en individuos con UPD, podría explicar la conducta obsesivo-compulsiva, así como podría desempeñar un papel en las habilidades cognitivas disminuidas(41).
Correlación genotipo-fenotipo en el síndrome de Angelman
La correlación genotipo-fenotipo entre los distintos mecanismos moleculares ha demostrado que los pacientes portadores de deleción muestran un fenotipo más grave en todos los aspectos del desarrollo neurológico(42-46). Manifiestan: una mayor tasa de epilepsia (90%, comparado con 75% en pacientes sin deleción), un inicio más temprano de convulsiones (media de edad 1,9 años, comparado con 6,3 años entre los individuos sin deleción) y pueden presentar un fenotipo de electroencefalograma más grave en comparación a las otras etiologías(47). Este grupo puede presentar hipopigmentación y una mayor frecuencia de microcefalia y dificultades motoras.
En cambio, los niños con SA debido a una mutación en UBE3A o a una disomia uniparental paterna, presentan habilidades lingüísticas significativamente mejores que los pacientes con deleción, en particular en el área expresiva, donde 6 de cada 9 niños con mutación en UBE3A o con disomía uniparental paterna pueden usar de 2 a 7 palabras, mientras que en el caso de los pacientes con deleción solo 3 de 30 niños(48). Se ha sugerido que los pacientes con SA portadores de mutaciones en UBE3A muestran un fenotipo intermedio entre los portadores de deleción y los portadores de disomía uniparental paterna. Presentan mayor incidencia de convulsiones y microcefalia, similar a los pacientes con deleción, mientras que su retraso del neurodesarrollo es similar a la disomía uniparental y al defecto de impronta(43,44).
Finalmente, los pacientes con disomía uniparental paterna y defecto de impronta tienen mejor crecimiento físico, tienen menos anomalías en el movimiento, y tienen una menor prevalencia (aunque no ausencia) de convulsiones, respecto a los pacientes con deleción(43).
Bibliografía
Los asteriscos reflejan el interés del artículo a juicio del autor.
1. Hagerman RJ. Angelman Syndrome and Prader-Willi Syndrome. En: Hagerman RJ. Neurodevelopmental Disorders. Diagnosis and Treatment. Oxford University Press; 1999. p. 243-90.
2. Cassidy SB, McCandless SE. Prader-Willi Syndrome. En: Cassidy SB, Allanson JE. Management of Genetic Syndromes. Wiley-Liss; 2005. p. 429-48.
3. Williams CA. Angelman Syndrome. En: Cassidy SB, Allanson JE. Management of Genetic Syndromes. Wiley-Liss; 2005. p. 53-62.
4.*** Gabau E, Baena N, Caixàs A, Novell R, Guitart M. Síndrome de Prader-Willi. En: Pérez de Nanclares G, Lapunzina P. Enfermedades de Impronta. Guías de buena práctica clínica; 2015. p. 147-80. ISBN978-84-608-2142-7.
5.*** Guitart M, Camprubí C, Fernández C, Gener B, Gabau E. Síndrome de Angelman. En: Pérez de Nanclares G, Lapunzina P. Enfermedades de Impronta. Guías de buena práctica clínica; 2015. p. 113-46. ISBN978-84-608-2142-7.
6. Bonello D, Camilleri F, Calleja-Agius. Angelman Syndrome: Identification and Management. Neonatal Network. 2017; 36(3): 142-50.
7. González-Lamuño Leguina D. Una visión general sobre las enfermedades raras. Pediatr Integral. 2014; XVIII(8): 550-63.
8. Abdilla Y, Barbara MA, Calleja-Agius J. Prader-Willi Syndrome: Background and Management. Neonatal Network. 2017; 36: 134-41.
9. Angulo MA, Butler MG, Cataletto ME. Prader-Willi síndrome: a review of clinical, genetic, and endocrine findings. J Endocrinol Invest. 2015; 38: 1249-63.
10. Passone CBG, Pasqualucci PL, Franco RR, Ito SS, Mattar LBF, Koiffmann CP, et al. Prader-Willi síndrome: What is the general Pediatrician supposed to do? – A review. Rev Paul Pediatr. 2018; 36: 345-52.
11. Heksch R, Kamboj M, Anglin K, Obrynba K. Review of Prader-Willi síndrome: the endocrine apprroach. Transl Pediatr. 2017; 6: 274-85.
12. Duis J, van Wattum PJ, Scheimann A, Salehi P, Brokamp E, Fairbrother l, et al. A multidisciplinary approch to the clinical management of Prader-Willi síndrome. Mol Genet Genomic Med; 2019. p. 1-21.
13. Wheeler A, Sacco P, Cabo R. Unmet clinical needs and burden in Angelman síndrome: a review of literatura. Orphanet Journal of Rare Diseases. 2017; 12: 164.
14.*** Gabau Vila E, Brun Gasca C, Fernández Zurita C, Joga Elvira, L, Ramírez Mallafré A. Análisis de los Síndromes de Angelman y afines, como los de Pitt Hopkins, de Mowat-Wilson, de Kleesfstra y de Phelan-McDermid. En McGrawHill education. Disability and Communication. Scientific Analysis, Total Comunication, ICT tolos and Case Studies; 2018. p. 1-22. ISBN: 978-84-486-1444-7.
15. Aguilera C, Gabau E, Laurie S, Baena N, Derdak S, Capdevila N, et al. Identification of a de novo splicing variant in the Coffin-Siris gene, SMARCE1, in a patient with Angelman-like síndrome. Mol Genet Genomic Med. 2019; 7: e00511.
16. Margolis SS, Gabrielle LS, Zbinden MA, Bird LM. Angelman Syndrome. Neurotherapeutics. 2015; 12: 641-50.
17. Chai J-H, Locke DP, Greally JM, Knoll JHM, Ohta T, Dunai J, et al. Identification of four highly conserved genes between breakpoint hotspots BP1 and BP2 of the Prader-Willi/Angelman syndromes deletion region that have undergone evolutionary transposition mediated by flanking duplicons. Am J Hum Genet. 2003; 73: 898-925.
18. Chamberlain SJ, Lalande M. Neurodevelopmental disorders involving genomic imprinting at human chromosome 15q11-q13. Neurobiol Dis. 2010; 39: 13-20.
19. Buiting K. Prader-Willi syndrome and Angelman syndrome. Am J Med Genet Part C Semin Med Genet. 2010; 154: 365-76.
20. Dagli A., Buiting K, Williams CA. Molecular and clinical aspects of Angelman syndrome. Mol Syndromol. 2012; 2: 100-12.
21. Sahoo T, Bacino CA, German JR, Shaw CA, Bird LM, Kimonis V, et al. Identification of novel deletions of 15q11q13 in Angelman syndrome by array-CGH: molecular characterization and genotype-phenotype correlations. Eur J Hum Genet. England; 2007; 15: 943-9.
22. Kim S-J, Miller JL, Kuipers PJ, German JR, Beaudet AL, Sahoo T, et al. Unique and atypical deletions in Prader-Willi syndrome reveal distinct phenotypes. Eur J Hum Genet. England. 2012; 20: 283-90.
23. Buiting K. Prader-Willi syndrome and Angelman syndrome. Am J Med Genet Part C Semin Med Genet. 2010; 154: 365-76.
24. Robinson WP, Christian SL, Kuchinka BD, Penaherrera MS, Das S, Schuffenhauer S, et al. Somatic segregation errors predominantly contribute to the gain or loss of a paternal chromosome leading to uniparental disomy for chromosome 15. Clin Genet. Denmark. 2000; 57: 349-58.
25. Robinson WP, Langlois S, Schuffenhauer S, Horsthemke B, Michaelis RC, Christian S, et al. Cytogenetic and age-dependent risk factors associated with uniparental disomy 15. Prenat Diagn. England. 1996; 16: 837-44.
26. Robinson WP, Wagstaff J, Bernasconi F, Baccichetti C, Artifoni L, Franzoni E, et al. Uniparental disomy explains the occurrence of the Angelman or Prader-Willi syndrome in patients with an additional small inv dup(15) chromosome. J Med Genet. England. 1993; 30: 756-60.
27. Buiting K, Gross S, Lich C, Gillessen-Kaesbach G, el-Maarri O, Horsthemke B. Epimutations in Prader-Willi and Angelman syndromes: a molecular study of 136 patients with an imprinting defect. Am J Hum Genet. United States. 2003; 72: 571-7.
28. Horsthemke B, Buiting K. Genomic imprinting and imprinting defects in humans. Adv Genet. United States; 2008; 61: 225-46.
29. Buiting K, Barnicoat A, Lich C, Pembrey M, Malcolm S, Horsthemke B. Disruption of the bipartite imprinting center in a family with Angelman syndrome. Am J Hum Genet. United States; 2001; 68: 1290-4.
30. Aguilera C, Vinas-Jornet M, Baena N, Gabau E, Fernandez C, Capdevila N, et al. Novel intragenic deletions within the UBE3A gene in two unrelated patients with Angelman syndrome: case report and review of the literature. BMC Med Genet. England. 2017; 18: 137.
31. Tan WH, Bird LM, Thibert RL, Williams CA. If not Angelman, what is it? A review of Angelman-like syndromes. Am J Med Genet Part A. 2014; 164: 975-92.
32. Ramsden SC, Clayton-Smith J, Birch R, Buiting K. Practice guidelines for the molecular analysis of Prader-Willi and Angelman syndromes. BMC Med Genet; 2010. p. 11:70.
33. Beckmann JS, Tomfohrde J, Barnes RI, Williams M, Broux O, Richard I, et al. A linkage map of human chromosome 15 with an average resolution of 2 cM and containing 55 polymorphic microsatellites. Hum Mol Genet. 1993; 2: 2019-30.
34. Mutirangura A, Greenberg F, Butler MG, Malcolm S, Nicholls RD, Chakravarti A, et al. Multiplex PCR of three dinucleotide repeats in the Prader-Willi/Angelman critical region (15q11-q13): molecular diagnosis and mechanism of uniparental disomy. Hum Mol Genet. 1993; 2: 143-51.
35. Driscoll DJ, Miller JL, Schwartz S, Cassidy SB. Prader-Willi Syndrome [Internet]. GeneReviews®. University of Washington, Seattle; 2017 [citado el 7 de marzo de 2019]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/20301505.
36. Poyatos D, Camprubí C, Gabau E, Nosas R, Villatoro S, Coll MD, et al. [Prader Willi syndrome patients: study of 77 patients]. Med Clin (Barc). 2009; 133: 649-56.
37. Hartley SL, Maclean WE Jr, Butler MG, Zarcone J, Thompson T. Maladaptive behaviors and risk factors among the genetic subtypes of Prader-Willi syndrome. Am J Med Genet A. 2005; 136: 140-5.
38. Cassidy SB, Forsythe M, Heeger S, Nicholls RD, Schork N, Benn P, et al. Comparison of phenotype between patients with Prader-Willi syndrome due to deletion 15q and uniparental disomy 15. Am J Med Genet. 1997; 68: 433-40.
39. Whittington J, Holland A, Webb T. Relationship between the IQ of people with Prader-Willi syndrome and that of their siblings: evidence for imprinted gene effects. J Intellect Disabil Res. 2009; 53: 411-8.
40. Yang L, Zhan GD, Ding JJ, Wang HJ, Ma D, Huang GY, et al. Psychiatric illness and intellectual disability in the Prader-Willi syndrome with different molecular defects—a meta analysis. PLoS One. 2013; 8: e72640.
41. Honea RA, Holsen LM, Lepping RJ, Perea R, Butler MG, Brooks WM, et al. The neuroanatomy of genetic subtype differences in Prader-Willi syndrome. Am J Med Genet B Neuropsychiatr Genet. 2012; 159B: 243-53.
42. Gentile JK, Tan W-H, Horowitz LT, Bacino CA, Skinner SA, Barbieri-Welge R, et al. A neurodevelopmental survey of Angelman syndrome with genotype-phenotype correlations. Journal of Developmental & Behavioral Pediatrics. 2010; 31: 592-601.
43. Lossie A, Whitney M, Ammidon D, Dong H, Chen P. Distinct phenotypes distinguish the molecular classes of Angelman syndrome. J Med Genet. 2001; 38: 834-45.
44. Moncla A, Malzac P, Voelckel M-A, Auquier P, Girardot L, Mattei M-G, et al. Phenotype-genotype correlation in 20 deletion and 20 non-deletion Angelman syndrome patients. Eur J Hum Genet. 1999; 7: 131-9.
45. Varela MC, Kok F, Otto PA, Koiffmann CP. Phenotypic variability in Angelman syndrome: comparison among different deletion classes and between deletion and UPD subjects. Eur J Hum Genet. 2004; 12(12): 987-92.
46. Sahoo T, Bacino CA, German JR, Shaw CA, Bird LM, Kimonis V, et al. Identification of novel deletions of 15q11q13 in Angelman syndrome by array-CGH: molecular characterization and genotype-phenotype correlations. Eur J Hum Genet. 2007; 15: 943-9.
47. Thibert RL, Larson AM, Hsieh DT, Raby AR, Thiele EA. Neurologic manifestations of Angelman syndrome. Pediatr Neurol. 2013; 48: 271-9.
48. Mertz LGB, Thaulov P, Trillingsgaard A, Christensen R, Vogel I, Hertz JM, et al. Neurodevelopmental outcome in Angelman syndrome: genotype-phenotype correlations. Res Dev Disabil. 2014; 35: 1742-7.
Bibliografía recomendada
- Enfermedades de Impronta. Guías de buena práctica clínica. Editado por Guiomar Pérez de Nanclares, Pablo Lapunzina. 2015. ISBN978-84-608-2142-7.
Revisión extensa de diferentes síndromes debidos a alteraciones de la impronta genómica. Libro gratuito.
- Duis J, van Wattum PJ, Scheimann A, Salehi P, Brokamp E, Fairbrother l, et al. A multidisciplinary approch to the clinical management of Prader-Willi síndrome. Mol Genet Genomic Med; 2019. p. 1-21.
El SPW es un trastorno multisistémico que requiere de un manejo multidisciplinar, la intervención médica y conductual ha mejorado el pronóstico de los pacientes. Revisión muy completa.
- Disability and Communication. Scientific Analysis, Total Comunication, ICT tools and Case Studies. En McGraw-Hill education. Encarnación Postigo Pinazo, Marina Calleja Reina, Elisabeth Gabau Vila. 2018. ISBN: 978-84-486-1444-7.
Culminación de un proyecto europeo Enhancing Communication (EC+) de las universidades de Gante, Klagenfurt, Málaga y del Hospital Universitario Parc Taulí. En que sobre diferentes entidades en las que hay una grave afectación del lenguaje oral, se revisan recursos de comunicación para los afectados, pero también para profesionales que deben atenderles en alguna ocasión. Libro gratuito.
| Caso clínico 1 |
|
Ingresa recién nacida de 24 horas de vida, procedente de la maternidad, por hipotonía y quejido respiratorio.
Madre: 32 años, primer embarazo de 39 SG, que ha cursado sin incidencias, con triple screening o cribado combinado de primer trimestre de bajo riesgo y ecografías normales. Diabetes gestacional.
Cesárea electiva por presentación podálica. Apgar 9-10-10. A la exploración física, se detectan edemas en manos y pies con acrocianosis, tono muscular bajo axial y de cintura escapular. Leve retrognatia. Se traslada a neonatología al persistir la hipotonía y presentar quejido respiratorio.
Peso: 3.030 g (P50); Talla: 49 cm (P25); PC: 36 cm (P90).
Evolución: inicialmente, precisa cánulas nasales; dada la persistencia de las necesidades de oxigenoterapia, se inicia VMNI (IPAP 14 y EPAP 6 que después se cambia a IPAP de 12 y EPAP de 5) con buena adaptación, y se puede retirar. Se realiza fisioterapia respiratoria durante el ingreso.
Buena tolerancia enteral con el biberón, pero con succión débil que va mejorando. Buen aumento ponderal.
Se da de alta a los 29 días de vida, con una exploración física que muestra: estrechez bifrontal, ojos almendrados, hipoactiva, pero reactiva a la manipulación, se cansa. Se despierta al estimularla, se consigue seguimiento ocular, pero se cansa y vuelve a cerrar los ojos, somnolencia. Tono axial bajo, hipotonía. Reflejo de Moro débil, pero presente.
Entre las pruebas complementarias destacamos:
• Ecografía cerebral: normal.
• Ecografía de caderas: displasia acetabular bilateral por inmadurez.
|
|
|
| Caso clínico 2 |
|
Visitamos a la Sra. Alicia de 42 años, con el diagnóstico de Discapacidad Intelectual Grave, a petición de la familia y de los profesionales que la atienden en el centro para discapacitados donde reside. El equipo médico del centro apunta que podría tratarse, por la clínica, de un caso de síndrome de Angelman.
Los padres refieren que Alicia era una lactante muy llorona, no diferenciaba el día de la noche. Presento crisis epilépticas del tipo ausencias. Adquirió la marcha libre a los 3,5 años; en lo referente al lenguaje, de pequeña decía “papá” y ahora “yaya”, siempre conducta hipermotórica, incluso ahora. Aspecto feliz. Siempre problemas para dormir por la noche. Requirió escolarización en educación especial.
A la exploración física, destacamos: piel clara, cráneo pequeño (PC: -1,5DS), braquicefalia, se muerde las uñas, hipermotórica.
|
|