|
| Temas de FC |
M.T. Muñoz Calvo
Servicio de Endocrinología. Hospital Infantil Universitario Niño Jesús. Departamento de Pediatría. Universidad Autónoma. Madrid
| Resumen
Para prevenir las enfermedades cardiovasculares en la vida adulta, es necesario identificar y tratar lo antes posible la hipercolesterolemia en los niños y en los adolescentes. Se debe realizar estudio a la población infantil de riesgo después de los 2 años, si existe historia familiar positiva o presencia de factores de riesgo cardiovascular en el niño. Las causas secundarias de hipercolesterolemias deben ser descartadas. El objetivo primordial del tratamiento dietético de las hipercolesterolemias será lograr que los niveles de C-LDL disminuyan, consiguiendo un descenso de un 10 a un 15%, aunque existen grandes variaciones individuales. Las estatinas deben ser incluidas entre los potenciales fármacos de primera línea por la experiencia adquirida en los últimos años, y por su capacidad de disminuir los niveles de C-LDL en torno a 18-45%, y sin afectación del crecimiento ni del desarrollo puberal. La edad de inicio del tratamiento con estatinas debe ser a partir de los 10 años, preferentemente a partir del Tanner II en los varones y después de la menarquia en las niñas. Otras opciones farmacológicas son el ezetimibe o las resinas de intercambio iónico |
| Abstract
To prevent cardiovascular diseases in adulthood, it is necessary to identify and treat as soon as possible hypercholesterolemia in children and adolescents. The study should be performed to child population at risk after two years, if there is a positive family history or presence of cardiovascular risk factors. Secondary causes of hypercholesterolemia should be discarded. The primary objective of the dietary treatment of hypercholesterolemia is to decrease LDL-C levels, achieving a decrease of 10 to 15%, although there are large individual variations. Statins should be included among potential first-line drugs, as per experience gained in recent years, and its ability to lower LDL-C levels around 18-45%, without affecting the growth and pubertal development. Treatment with statins should be started at 10 years of age and over, preferably starting with TannerII in males and after menarche in girls. Other pharmacological options include ezetimibe or ion exchange resins |
Palabras clave: Colesterol; Triglicérido; Dieta; Estatinas y Fibratos
Key words: Cholesterol; Triglyceride; Diet; Statins and Fibrates
Pediatr Integral 2015; XIX (5): 355-364
Dislipemias
Desarrollo de aterosclerosis en los niños
El desarrollo de la aterosclerosis es un proceso que se inicia en la infancia y que progresa con una velocidad que depende de la presencia o ausencia de determinados factores de riesgo cardiovascular (FRCV), como han demostrado diferentes estudios(1). Las estrías grasas y placas fibrosas estaban presentes en casi el 50% y 8%, respectivamente, de los individuos en la infancia, y en casi el 70% y 5% de adultos jóvenes, y la extensión de estas lesiones ateroscleróticas se correlacionaban con los niveles de colesterol total (CT), colesterol unido a las lipoproteínas de baja densidad (C-LDL) y triglicéridos(2).
Más recientemente, los métodos incruentos por la imagen han permitido el estudio del desarrollo de la aterosclerosis. Un estudio realizado por ecografía de las arterias carótidas, para evaluar el espesor de la íntima y la media, que es un indicador del proceso aterosclerótico en los adultos, demostró que el aumento de espesor de la íntima y la media se asociaba con incrementos del CT y otros FRCV, como la hipertensión arterial en la infancia(3). La alteración de la función endotelial ha sido descrita en niños con hiperlipemias y está relacionada con la fisiopatología de la aterosclerosis.
Las concentraciones plasmáticas de CT reflejan una situación compleja, con múltiples dependencias: las de origen genético, las derivadas de la situación ambiental (tipo de dieta y grado de ejercicio físico) y las consideradas poligénicas, que dependen de la interacción de algunos determinantes genéticos con la influencia derivada de los dietéticos(4). Los factores genéticos son poco modificables y los dietéticos muchas veces tienen su origen en la infancia; por lo tanto, una educación adecuada podría desempeñar un papel preventivo sobre el desarrollo futuro de enfermedades cardiovasculares (ECV).
Niveles de lipoproteínas en la edad pediátrica
Los niveles plasmáticos de lipoproteínas son diferentes en la infancia y adolescencia en comparación con la edad adulta. Los niveles de CT, C-LDL, colesterol unido a proteínas de alta densidad (C-HDL) y triglicéridos (TG) ascienden paulatinamente desde el nacimiento y se estabilizan entre los 2 y 4 años, manteniéndose en un mismo percentil a lo largo del tiempo durante los años prepuberales (fenómeno tracking del colesterol)(5). A partir de los 10-12 años, los niveles plasmáticos de CT y C-LDL disminuyen entre un 5 y un 10% en ambos sexos –es más evidente en varones–, debido al descenso acusado del C-HDL, pero en los últimos años de la adolescencia, se produce un reascenso de CT y C-LDL alcanzando niveles medios de adulto a partir de los 20 años. En valores absolutos, de los 13 a los 19 años, el CT es consistentemente más alto en mujeres que en varones. La disminución de los niveles de C-HDL, es decir, patrón más aterogénico, que se produce durante la pubertad en los varones es el cambio lipídico más importante y va a permanecer durante la etapa adulta(5).
Clasificación de las dislipemias
Las dislipemias pueden ser clasificadas en primarias y secundarias. Se denominan primarias (Tabla I), cuando hay factores genéticos implicados en su etiología. Según los genes implicados, las hiperlipemias pueden ser monogénicas, producidas por la alteración de un solo gen responsable del metabolismo lipídico, o poligénicas, si están producidas por la suma de los efectos de diferentes genes(6). Estas últimas, son más frecuentes y menos severas que las monogénicas, y su expresión fenotípica está influenciada por factores ambientales (dieta, ejercicio…) (Tabla I). El diagnóstico de dislipemia primaria exige haber descartado antes posibles causas de dislipemia secundaria, la mayoría de las cuales suelen evolucionar favorablemente con el tratamiento de la enfermedad causal.

Dislipemias primarias
Hipercolesterolemia familiar monogénica
Actualmente, se conocen, al menos, tres formas diferentes: la clásica, otras mutaciones en proteínas distintas del receptor de LDL con herencia autosómica dominante, y la hipercolesterolemia con herencia autosómica recesiva.
Forma clásica: está causada por mutaciones en el gen del receptor LDL (LDLR, 19p13.2), produciendo aumento de las concentraciones en sangre de C-LDL, ya desde edades precoces. Esta mutación puede heredarse en homocigosis o en heterocigosis. Si se hereda en homocigosis, los síntomas aparecen antes de los 10 años de edad. Los hallazgos característicos son: los xantomas (tendinosos y cutáneos), el arco corneal y el aumento de CT (en torno a 1.000 mg/dl) y de C-LDL (mayor de 600 mg/dl)(7). Todo ello conlleva al desarrollo de ateroesclerosis generalizada de forma precoz. Si la mutación se hereda en heterocigosis, la aparición de los síntomas y alteraciones lipídicas es más tardía y más leve. La aparición de xantomas está en relación con la duración e intensidad de la hipercolesterolemia, siendo infrecuentes antes de los 20 años. Los valores de CT se encuentran entre 270 y 550 mg/dl y los de LDL-c son superiores a 160 mg/dl. En España, se han descrito más de 400 mutaciones en el gen del receptor de LDL asociadas a hipercolesterolemia familiar (HF); sin embargo, la tasa de detección de una mutación funcional en casos con diagnóstico clínico varía del 20 al 80%, por tanto, un test genético negativo no excluye el diagnóstico, sobre todo, cuando el fenotipo sugiere una HF(7).
Actualmente, se puede realizar diagnóstico genético de esta entidad caracterizando la mutación del gen del receptor del LDL.
Otras hipercolesterolemias monogénicas con herencia autosómica dominante: son debidas a mutaciones en el gen de la ApoB (apoB100; 2p24) y a una mutación de la proproteína convertasa subtilisina/kexina tipo 9 (PCSK9, 1p34.1-p32). Esta proteína está implicada en la degradación del receptor de LDL y su alteración produce una disminución del aclaramiento de las LDL.
Hipercolesterolemia autosómica recesiva (ARH): es debida al déficit del adaptador de proteína ARH o a la incapacidad del mismo para interactuar con el receptor de LDL; por ello, cursa con disminución del aclaramiento de LDL y aumento de LDL-c y CT. Su fenotipo es similar a la hipercolesterolemia familiar homocigota, pero, en general, menos severo y con mayor respuesta a los fármacos hipolipemiantes. Cursa con xantomas grandes desde la infancia, CT mayor de 500 mg/dl, y aparición de ECV en menores de 30 años(7).
Hipercolesterolemia familiar homocigota
Es una forma rara de HF que se produce cuando se hereda la misma mutación en el gen del RLDL de ambos progenitores. Se estima que afecta a 1 caso por cada 800.000 a un millón de personas, y los pacientes presentan hipercolesterolemia grave, xantomas y ateroesclerosis acelerada. El diagnóstico se debe realizar alrededor de los 2 años o, inclusive antes, y se basa en una concentración de C-LDL sin tratamiento >500 mg/dL o C-LDL con tratamiento máximo >300 mg/dL, presencia de xantomas antes de los 10 años e historia de hipercolesterolemia o de diagnóstico genético en ambos progenitores(8).
Hiperlipemia familiar combinada (HFC)
Se caracteriza por una síntesis hepática aumentada de apo B-100, lo que da lugar a una sobreproducción hepática de C-VLDL. Se manifiesta a partir de la 2ª década de la vida, con aumento de CT (250-500 mg/dl) y/o TG (250-750 mg/dl). El diagnóstico se basa en la historia familiar y en la presencia de varias anomalías lipídicas. No existe diagnóstico genético definitivo en la actualidad(9).
Hipercolesterolemia poligénica
Su etiología es desconocida, se piensa que ocurre por alteraciones en diversos genes reguladores del metabolismo del colesterol. Las combinaciones de esta herencia poligénica junto con factores ambientales, especialmente la dieta, darían lugar al aumento del colesterol. Es la forma más frecuente de hipercolesterolemia primaria en la población general, representando el 80% de todas ellas(9). Clínicamente, se presenta con aumentos moderados de colesterol, con valores entre el percentil 75-95, con padres y hermanos sanos o con elevaciones de colesterol similares. El diagnóstico es clínico, una vez excluidas la hipercolesterolemia familiar y la hiperlipemia familiar combinada.
Hipertrigliceridemia familiar
Se caracteriza por el aumento de la síntesis hepática de VLDL. Se han descrito varias alteraciones, entre ellas, el déficit de lipoproteín-lipasa y de ApoCII. Cursa con aumento de TG (200-500 mg/dl) a expensas de la fracción de colesterol ligado a lipoproteínas de muy baja densidad (C-VLDL), o con aumento de C-VLDL, TG y quilomicrones. La clínica es variable dependiendo del grado de hipertrigliceridemia. Su diagnóstico es clínico(6).
Déficit familiar de apo B-100
La alteración se debe a una apolipoproteína B-100 defectuosa, que no se une al receptor de LDL, elevándose el C-LDL en suero. Hasta ahora, se han identificado 3 mutaciones que dan lugar al déficit familiar de apo B-100(6). El diagnóstico es genético por técnicas de biología molecular.
Beta-sitosterolemia
Se produce por mutación del trasportador ABCG5/ABCG8, implicado en el transporte del colesterol no esterificado por las células epiteliales intestinales hacia el lumen del intestino(10). Cursa con aumento de esteroles vegetales y C-LDL, xantomatosis tendinosa y xantelasmas. Tienen riesgo de enfermedad cardiovascular prematura.
Hipoalfalipoproteinemia
Se conoce la existencia de varios defectos genéticos infrecuentes que pueden producir la reducción de los niveles de C-HDL en plasma. La mutación en el gen que codifica el transportador ABCA1 puede producir varias formas clínicas. La más grave es la enfermedad de Tangier, caracterizada por la casi ausencia de C-HDL (menor de 5 mg/dl) en plasma y la acumulación de ésteres de colesterol en tejidos, como: hígado, bazo, ganglios linfáticos, amígdalas, timo, mucosa intestinal, nervios periféricos y córnea. A pesar de que los niños con enfermedad de Tangier tienen bajos niveles de C-LDL, se ha observado que presentan un riesgo elevado de sufrir ECV durante la edad adulta. La deficiencia de lecitil-colesterol-aciltransferasa (LCAT) y la deficiencia de apolipoproteína A1 (apoA1) son otras causas de disminución de C-HDL(6).
En las formas más leves de hipoalfalipoproteinemia (C-HDL entre 15-35 mg/dl), la expresividad clínica es variable, pudiendo ser causa de enfermedad cardiovascular prematura. No existe un consenso respecto al tratamiento, dado que no hay experiencia con fármacos modificadores de C-HDL en niños(10). Es por ello que, el tratamiento con fármacos que disminuyen la relación C-LDL/C-HDL se inicia más tardíamente. En el caso de la hipoalfalipoproteinemia leve, la única medida es aconsejar una dieta cardiosaludable(6).
Dislipemias secundarias
A continuación, se enumeran las principales etiologías de dislipemia secundaria agrupadas según la alteración lipídica con la que cursen predominantemente(11):
• Hipercolesterolemia: porfiria aguda intermitente, ingesta elevada de ácidos grasos saturados, anorexia nerviosa.
• Hipertrigliceridemia: síndrome de Cushing, lipodistrofia, glucogenosis tipo 1, nefritis lúpica, terapia con retinoides.
• Aumento de CT y TG: trasplante de órgano sólido.
• Disminución de C-HDL: obesidad, vida sedentaria, hábito tabáquico.
• Aumento de CT y C-LDL: hipotiroidismo, síndrome nefrótico, tratamiento antirretroviral (VIH), enfermedad obstructiva hepática.
• Aumento de TG y C-VLDL: obesidad, síndrome metabólico, diabetes mellitus tipo 2, insuficiencia renal crónica, terapia con corticoides, hipotiroidismo, anticonceptivos orales, embarazo, ingesta de alcohol.
Orientación diagnóstica
Se considera dislipemia a los valores por encima del percentil 95 para la edad y sexo de CT, C-LDL y triglicéridos (TG), y los valores de C-HDL por debajo del percentil 10. El NCEP (National Cholesterol Education Program) pediátrico(12) propone unos valores deseables, en el límite y de riesgo (Tabla II).

El objetivo principal de diagnosticar una hipercolesterolemia en la infancia es el inicio de un tratamiento eficaz, especialmente la instauración de hábitos de vida saludables lo antes posible, para prevenir el desarrollo de ECV que se manifiesta en la edad adulta(5).
Cribado de las dislipemias
La práctica del cribado de las dislipemias en niños o adolescentes es un tema controvertido, en lo que respecta a quién y cuándo hacerlo, debido a los riesgos desconocidos de un tratamiento hipolipemiante a largo plazo iniciado en edades tempranas. Existen diferentes estrategias para la detección en los niños, como son: el cribado en cascada familiar, el cribado universal y el cribado selectivo basado en la historia familiar. En España y en otros países europeos, el cribado en los niños se suele hacer como parte del cribado en cascada familiar, cuando se conoce el diagnóstico en la familia. El cribado selectivo debe realizarse a la población infanto-juvenil de riesgo entre los 2 y los 8 años, y entre los 12 y los 16 años(5). Se considera población de riesgo, si existe historia familiar positiva o factores de riesgo en el niño:
Historia familiar
• ECV prematura en padres, abuelos o tíos (varones < 55 años o mujeres < 65 años).
• Padres con dislipemia conocida o CT mayor o igual a 240 mg/dl.
Factores de alto riesgo
• Hipertensión arterial que requiere tratamiento farmacológico (TA > P 99 + 5 mmHg).
• Hábito tabáquico.
• IMC ≥ percentil 97.
• Presencia de condiciones de alto riesgo: diabetes mellitus, enfermedad renal crónica, trasplante de órganos, enfermedad de Kawasaki con aneurismas.
Factores de riesgo moderado
• Hipertensión arterial que no requiere tratamiento farmacológico.
• IMC ≥ percentil 95 y < percentil 97.
• C-HDL< 40 mg/dl.
• Presencia de condiciones de riesgo moderado: enfermedad de Kawasaki con aneurismas en regresión, enfermedad inflamatoria crónica (lupus eritematoso, artritis idiopática juvenil), VIH y síndrome nefrótico.
Diagnóstico
El estudio analítico debe efectuarse en las siguientes condiciones:
1. Ayuno de 12 horas.
2. Sin modificaciones de la dieta habitual.
3. Libre de enfermedad desde varias semanas antes.
4. Teniendo en cuenta la toma de ciertos fármacos que pueden modificar los lípidos.
En caso de valores lipídicos alterados, se repetirá el estudio analítico tres o cuatro semanas después, para confirmar la alteración de la analítica(13).
Se deberá realizar una historia familiar (antecedentes de ECV prematura en padres o abuelos, antecedentes en padre o madre de dislipemia genética). Asimismo, conocer los antecedentes personales: hábitos alimentarios, estilos de vida, toma de fármacos, hábito tabáquico y se descartarán posibles causas de dislipemia secundaria.
En la exploración física, se anotarán las medidas antropométricas: peso, talla, IMC y estadio puberal, además de la toma de la tensión arterial.
En el estudio analítico, se solicitará:
• Hemograma, Bioquímica (perfil hepático y renal) y hormonas tiroideas.
• Perfil lipídico en ayunas: CT, TG y lipoproteínas (C-LDL, C-HDL y C-VLDL).
• Según sospecha etiológica: apolipoproteína A, B, y Lp(a).
Criterios diagnósticos de hipercolesterolemia familiar heterocigota (Tabla III):

• Confirmar la trasmisión vertical de la hipercolesterolemia y/o ECV prematura en uno de los padres.
• Niveles de C-LDL ≥ 190 mg/dl o bien niveles de C-LDL ≥ 150 mg/dl, cuando se tiene la confirmación genética de HF en uno de los padres.
• El estudio genético nos permitirá dar una información pronóstica a los pacientes y se realizará mediante(14):
- La demostración de mutaciones en el gen del receptor LDL (LDLR, 19p13.2). La tasa de detección de una mutación funcional en casos con diagnóstico clínico varía del 20 al 80%, por tanto, un test genético negativo no excluye el diagnóstico, sobre todo, cuando el fenotipo sugiere una HF.
- Otras causas menos frecuentes son las mutaciones en el gen de la ApoB (apoB100; 2p24) y en el gen de la proproteína convertasa subtilisina/kexina tipo 9 (PCSK9, 1p34.1-p32)(14).
- Como resultado de estos defectos moleculares se produce una disminución en la eliminación del colesterol plasmático unido a lipoproteínas de baja densidad, y esto conlleva a que estos pacientes presenten desde el nacimiento valores plasmáticos elevados de CT y C-LDL.
Tratamiento de las dislipemias (Fig. 1)
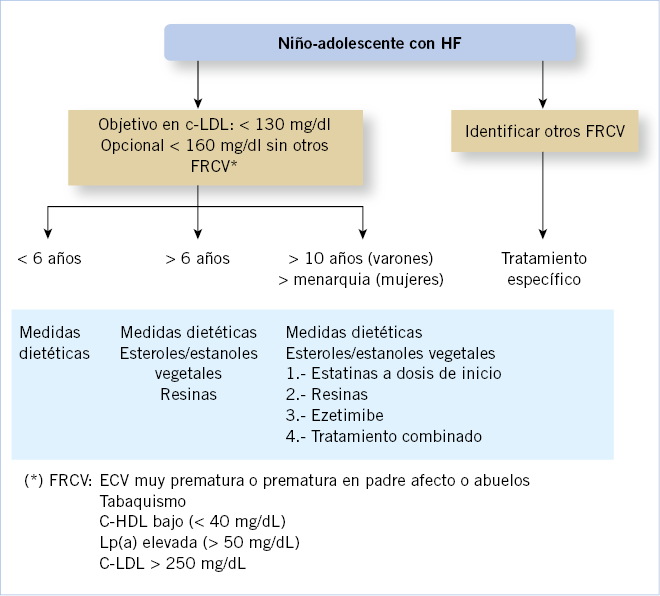
Figura 1. Algoritmo de tratamiento en niños y adolescentes con hipercolesterolemia familiar.
Recomendaciones dietéticas
Durante toda la infancia y adolescencia, es esencial un aporte adecuado de energía y nutrientes para que el crecimiento sea normal. Las modificaciones en la dieta deben introducirse de manera progresiva, lo cual será beneficioso para el niño y su familia.
El objetivo primordial del tratamiento dietético de las hipercolesterolemias será lograr que los niveles de C-LDL disminuyan, consiguiendo un descenso de un 10 a un 15%, aunque existen grandes variaciones individuales(15).
Las características de esta dieta están expuestas en la tabla IV.

Si después de 3 meses no se logran los objetivos terapéuticos con la dieta inicial, se introducirán otras modificaciones (Dieta 2): ingesta total de grasa 25-30% de las calorías diarias, de grasa saturada <7% de las calorías, de grasa monoinsaturada 10% y de colesterol <200 mg al día, orientada a reducir el C-LDL o los TG, siempre bajo la supervisión de un dietista. Las características de esta dieta se exponen en la figura 2. Si a pesar de su cumplimiento no se alcanzan los objetivos marcados, se deberá considerar el tratamiento farmacológico.

Figura 2. Recomendaciones dietéticas (Dieta 2): adaptado del Expert Panel on Integrated Guidelines for Cardiovascular Health and Risk Reduction in Children and Adolescents.
La disminución del aporte de grasas y colesterol que sufren con frecuencia los niños que presentan una dislipemia, puede tener repercusiones sobre su crecimiento. Se ha recomendado prudencia acerca de la utilización de dietas demasiado restrictivas; ya que, al disminuir la ingesta de grasa se corre el riesgo de no proporcionar suficiente energía para asegurar un óptimo crecimiento(16).
Actividad física
Los niños y los adolescentes deben dedicar un número establecido de horas al día al ejercicio físico en los colegios, promover la participación en actividades deportivas según la edad, y dieta cardiosaludable en los menús escolares(5).
La Academia Americana de Pediatría recomienda disminuir las actividades sedentarias (televisión, juegos de ordenador, etc.) a menos de 2 horas al día en los niños > 2 años.
Prevención primaria del consumo de tabaco y alcohol
Se ha demostrado el papel del tabaquismo como factor de riesgo cardiovascular, ya que se asocia a la aparición de dislipemia e insulinorresistencia, favorece un estado proinflamatorio e incrementa el estrés oxidativo celular(5).
Suplementos dietéticos
El aumento de la ingesta de fibra puede ser útil en la disminución de los niveles de C-LDL, en torno al 5-10%. La dosis adecuada (AAP) se calcula como la edad del niño + 5 g/día, hasta una dosis de 20 g/día a los 15 años de edad(17).
Los estanoles y esteroles de plantas se añaden a las margarinas, zumos de naranja, yogur líquido, barras de cereales y suplementos dietéticos. Son compuestos naturales cuya estructura química es similar al colesterol, actúan disminuyendo la absorción intestinal de colesterol exógeno de origen alimentario, así como del colesterol endógeno de origen biliar, demostrándose en adultos una disminución del C-LDL de un 10-20%, con mínimos efectos adversos(17).
Las proteínas de soja disminuyen los niveles de triglicéridos y C-VLDL e incrementan los niveles de C-HDL. Los suplementos de una dieta baja en grasa, enriquecida en ácidos grasos omega-3, con altas concentraciones de ácido eicosapentanoico y ácido docosahexanoico, han demostrado que incrementan significativamente las LDL grandes (menos aterogénicas) y disminuyen las LDL pequeñas (más aterogénicas). Es bien tolerado, con escasos efectos secundarios (gastrointestinales, hiperglucemia y elevaciones de las enzimas hepáticas)(17).
Tratamiento farmacológico
Estatinas
Las estatinas deben ser incluidas entre los potenciales fármacos de primera línea por la experiencia adquirida en los últimos años y por su capacidad de disminuir los niveles de C-LDL en torno a 18-45%, y sin afectación del crecimiento ni del desarrollo puberal(18,19).
Edad de inicio
No existe un consenso acerca a qué edad comenzar el tratamiento con estatinas. Diferentes Guías(5,20,21) recomiendan iniciar tratamiento en niños ≥ 10 años (preferiblemente en estadio II Tanner o superior en niños y a partir de la menarquia en niñas), tras 6-12 meses de dieta baja en grasas y colesterol, si:
• C-LDL ≥ 190 mg/dl.
• C-LDL entre 160 mg/dl y 189 mg/dl, con historia de ECV prematura en familiares de primer grado, o existe otros FRCV (tabaco, C-HDL bajo, Lp(a) elevada, entre otros).
• En la hipercolesterolemia familiar homocigota, el tratamiento debe ser iniciado en el momento del diagnóstico.
La pravastatina está aprobada por la FDA y la EMA en hipercolesterolemia primarias, a partir de los 8 años. La dosis recomendada es de 10 a 20 mg/día en niños de 8 a 13 años y de 10 a 40 mg/día desde los 14 a los 18 años. Otras estatinas aprobadas por la FDA son: lovastatina, simvastatina, atorvastatina, fluvastatina y rosuvastatina(22).
Dosificación
• Empezar con la dosis más baja una vez al día, generalmente al acostarse. Medir niveles basales de CPK, ALT y AST.
• La meta es conseguir niveles de LDL < 130 mg/dL.
• Tras 4 semanas de tratamiento, determinar: perfil de lipoproteínas en ayunas, CPK, ALT, AST. Repetir control analítico a las 8 semanas y a los 3 meses(22).
• Si hay anomalías de laboratorio o aparecen síntomas, suspender temporalmente el fármaco y repetir analítica en 2 semanas. Cuando los valores retornen a la normalidad, el fármaco puede reiniciarse con monitorización estrecha.
Seguimiento
• Valorar el ritmo de crecimiento y el desarrollo puberal (peso, talla, índice de masa corporal, estadio de Tanner).
• Monitorizar el perfil de lipoproteínas en ayunas, CPK, ALT y AST cada 3-6 meses(22).
• Insistir al paciente sobre medidas dietéticas e informar sobre otros factores de riesgo, tales como: sobrepeso, sedentarismo y tabaquismo, entre otros.
Eficacia y seguridad
• Se han publicado revisiones sobre su utilización en la infancia, concluyendo que a la vez de seguras, son muy eficaces.
• En cuanto a los efectos adversos, no se han publicado casos de miositis, miopatía o rabdomiolisis. Se han encontrado algunos casos de elevaciones asintomáticas de la CPK y enzimas hepáticas, que han revertido con un descenso de las dosis de la medicación, sin necesidad de suspender el tratamiento. Tampoco se han encontrado alteraciones del ritmo de crecimiento, desarrollo puberal ni del metabolismo de las vitaminas(23).
• Las estatinas están contraindicadas en el embarazo, por lo que debe advertirse a las adolescentes y enviarlas, en caso necesario, a consejo ginecológico.
En la figura 3, está representado el algoritmo terapéutico de las dislipemias, basado en el C-LDL.
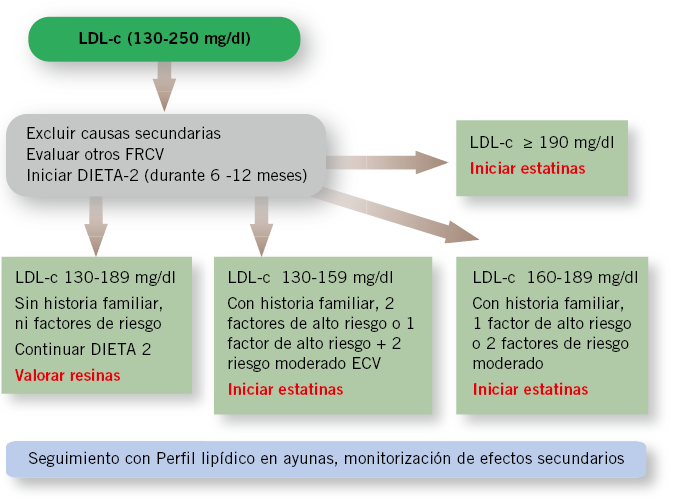
Figura 3. Algoritmo para abordar las dislipemias, basado en los niveles de C-LDL. Adaptado del Expert Panel on Integrated Guidelines for Cardiovascular Health and Risk Reduction in Children and Adolescents.
Resinas de intercambio iónico
• No se absorben y son seguras a largo plazo.
• Se pueden administrar a partir de los 6 años, sin historia familiar de enfermedad cardiovascular o factores de riesgo, si el C-LDL ≥ 130mg/dl y < 190mg/dl, tras 6-12 meses de dieta baja en grasas y colesterol(24).
• Sin embargo, por su baja palatabilidad y por sus efectos adversos a nivel gastrointestinal, como flatulencia y estreñimiento, han caído en desuso. Reducen los niveles de C-LDL de un 16% a un 19%(24).
• La dosis media recomendada es de 0,25-0,35 g/kg/día, con una dosis máxima de 8 g/día para el colestipol y de 10 g para la colesteramina. Se debe iniciar el tratamiento con dosis bajas, e incrementar progresivamente. El colesevelam, tiene menos efectos secundarios, aunque no existe aún experiencia en Pediatría(25).
Ezetimibe
Actúa inhibiendo selectivamente la absorción intestinal de colesterol (tanto dietético como de origen biliar) en el borde en cepillo de los enterocitos. En estudios preclínicos, no se ha observado que altere la absorción de triglicéridos, ácidos grasos, ácidos biliares, progesterona, etinilestradiol o vitaminas liposolubles A y D(26).
Los principales efectos adversos han sido gastrointestinales (diarrea y dolor abdominal) y cefalea. Reducen los niveles de CT y C-LDL, del 23% y 30%, respectivamente, a dosis de 10 mg/día.
Su uso está autorizado por la FDA y la EMA, a partir de los 10 años de edad. Sin embargo, no existen datos de seguridad a largo plazo en población pediátrica.
Otros fármacos
Fibratos
• Reducen los niveles de TG y elevan los niveles de C-HDL. Los principales efectos secundarios son mialgias, debilidad muscular e incremento de CPK, ALT y AST.
• Niveles de TG entre 200 y 499 mg/dl, realizar tratamiento dietético (Fig. 2), cambio de estilo de vida y aumento de ingesta de pescado (salmón, caballa, arenque [rango de 1,5-3 g/100 g de pescado]), y valorar tratamiento con suplementos de aceite de pescado (Omacor®, dosis inicial 1 g/día)(27).
• Valorar tratamiento con aceite de pescado o fibratos (bezafibrato, gemfibrozilo, fenofibrato) en niños con hipertrigliceridemia primaria y TG ≥ 500 mg/dl, tras 6-12 meses de tratamiento dietético (Fig. 2) y cambio de estilo de vida. En la figura 4, está representado el algoritmo terapéutico de las hipertrigliceridemias.

Figura 4. Algoritmo para abordar las dislipemias basado en niveles de TG. Adaptado deExpert Panel on Integrated Guidelines for Cardiovascular Health and Risk Reduction in Children and Adolescents.
Ácido nicotínico
Es el único fármaco que reduce los niveles de lipoproteína (a). Sin embargo, no puede ser recomendado en niños por su pobre tolerancia, la elevada frecuencia de efectos adversos (rubefacción, elevación de aminotransferasas, dolor abdominal, vómitos y cefalea) y los potenciales efectos secundarios graves (fallo hepático fulminante)(5).
Tratamientos emergentes
En los pacientes con HF homocigota o con HF heterocigota grave, el tratamiento combinado, utilizando estatinas potentes a dosis altas y ezetimibe, no es suficiente para reducir los niveles de C-LDL, persistiendo, por tanto, el elevado riesgo cardiovascular. Esto ha estimulado el desarrollo de terapias innovadoras que pueden proporcionar una reducción eficaz y mantenida del C-LDL, como son:
• Los inhibidores de la MTTP (microsomal triglyceride transfer protein). El lomitapide inhibe la enzima que se encarga de ensamblar los ácidos grasos libres con la Apo B en el hígado e intestino, disminuyendo la síntesis de VLDL y quilomicrones. La Agencia Europea y Española del Medicamento han aprobado su indicación en los pacientes con HF Homocigota mayores de 18 años(28).
• Los anticuerpos monoclonales anti PCSK9 basan su efecto en que esta proteína está relacionada con el reciclado del RLDL. Producen una reducción del C-LDL entre el 40 y 72% y, además, se ha observado una reducción en los niveles de Lp(a) por un mecanismo aún no conocido(29).
LDL-aféresis
La LDL aféresis es el tratamiento de elección para los pacientes con HF homocigota y ha demostrado tener un efecto beneficioso en la ateroesclerosis aórtica y coronaria, mejorando la supervivencia. Es una estrategia terapéutica segura y eficaz, en los que se ha demostrado la eliminación de xantomas, la regresión angiográfica de la aterosclerosis coronaria y la reducción de los episodios coronarios mortales y no mortales. La LDL-aféresis se puede comenzar a partir de los 6 años y siempre antes de los 10 años, por el elevado riesgo de estenosis aórtica grave(29).
Existen en la actualidad, diversos métodos de LDL-aféresis, siendo los más utilizados los sistemas de absorción con columnas de sulfato de dextrano y la absorción directa de lipoproteínas. Permite la eliminación específica de C-LDL y Lp (a) con una disminución plasmática del 50-75%, cuando se usa semanalmente o cada dos semanas. El tratamiento con estatinas se debe mantener para retrasar el efecto rebote en el aumento del C-LDL(30).
Recomendaciones para el Pediatra de Atención Primaria
1. Realizar estudio a la población infantil de riesgo después de los 2 años, si existe historia familiar positiva o presencia de factores de riesgo cardiovascular en el niño.
2. Las causas secundarias de hipercolesterolemias deben ser descartadas.
3. La edad y los niveles de C-LDL deben ser utilizados para realizar el diagnóstico de HF, siendo los niveles de C-LDL ≥ 190 mg/dl o C-LDL entre 160 mg/dl y 189 mg/dl con historia de ECV prematura en familiares de primer grado indicativos de alta probabilidad de HF (se recomiendan dos analíticas en ayunas).
4. Los pacientes deben ser clasificados en las diferentes categorías de riesgo, según la edad, presencia de otros FRCV, historia familiar de ECV prematura, y los niveles de hipercolesterolemia al diagnóstico.
5. Se deben realizar cambios en los estilos de vida (dieta, actividad física, hábito tabáquico).
6. La edad de inicio del tratamiento con estatinas debe ser a partir de los 10 años, preferentemente a partir del Tanner II en los varones y después de la menarquia en las niñas (tras 6-12 meses de dieta baja en grasas).
7. La pravastatina está aprobada por la EMA, a partir de los 8 años.
8. Los objetivos del tratamiento farmacológico son conseguir unos niveles de LDL: mínimo: <130 mg/dL, ideal: <110 mg/dL.
9. Es necesario monitorizar el crecimiento: peso, talla, índice de masa corporal y estadio de Tanner.
10. Control de CPK, ALT y AST al inicio y cada 3-6 meses del tratamiento con estatinas.
Bibliografía
1. Narverud I, Retterstøl K, Iversen PO, et al. Markers of atherosclerotic development in children with familial hypercholesterolemia: a literature review. Atherosclerosis. 2014; 2351: 299-309.
2. Kusters DM, Wiegman A, Kastelein JJ, Hutten BA. Carotid intima-media thickness in children with familial hypercholesterolemia. Circ Res. 2014; 114(2): 307-10.
3. Davis PH, Dawson JD, Riley WA, Lauer RM. Carotid intimal-medial thickness is related to cardiovascular risk factors measured from childhood through middle age: The Muscatine Study. Circulation. 2001; 104: 2815-9.
4. Gómez-Gerique JA, Gutiérrez-Fuentes JA, Montoya MT, et al. Lipid profile of the Spanish population: the DRECE (diet and risk of cardiovascular disease in Spain) study. DRECE study group. Med Clin (Barc). 1999; 113: 730-5.
5. Expert Panel on Integrated Guidelines for Cardiovascular Health and Risk Reduction in Children and Adolescents; National Heart, Lung, and Blood Institute. Expert panel on integrated guidelines for cardiovascular health and risk reduction in children and adolescents: summary report. Pediatrics. 2011; 128 Suppl 5: S213-56.
6. Bamba V. Update on screening, etiology, and treatment of dyslipidemia in children. J Clin Endocrinol Metab. 2014; 99: 3093-102.
7. Fellin R, Arca M, Zuliani G, Calandra S, Bertolini S. The history of Autosomal Recessive Hypercholesterolemia (ARH). From clinical observations to gene identification. Gene. 2015; 555: 23-32.
8. Huang CH, Chiu PC, Liu HC, et al. Clinical observations and treatment of pediatric homozygous familial hypercholesterolemiadue to a low-density lipoprotein receptor defect. J Clin Lipidol. 2015; 9: 234-40.
9. Gaddi A, Cicero AF, Odoo FO, Poli AA, Paoletti R; Atherosclerosis and Metabolic Diseases Study Group. Practical guidelines for familial combined hyperlipidemia diagnosis: an up-date. Vasc Health Risk Manag. 2007; 3: 877-86.
10. Lütjohann D, von Bergmann K. Phytosterolaemia: diagnosis, characterization and therapeutical approaches. Ann Med. 1997; 29: 181-4.
11. Kwiterovich P. Primary and secundary disorders of lipid metabolism in pediatrics. Pediatr Endocrinol Rev. 2008; 5: 727-38.
12. National Cholesterol Education Program. Report of the Expert Panel on Blood Cholesterol Levels in Children and Adolescents. Pediatrics. 1992; 89: 525-84.
13. Braamskamp MJ, Hutten BA, Wiegman A, Kastelein JJ. Management of hypercholesterolemia in children. Paediatr Drugs. 2014; 16(2): 105-14.
14. Vogt A. The genetics of familial hypercholesterolemia and emerging therapies. Appl Clin Genet. 2015; 28; 8: 27-36.
15. Morais A, Lama R, Dalmau S y Comité de Nutrición de la AEP. Hipercolesterolemia. Abordaje terapéutico. An Pediatr (Barc). 2009; 70: 488-96.
16. Muñoz MT, Barrios V, Pozo J, Argente J. Influencia del tratamiento dietético sobre el crecimiento en niños con dislipemias primarias. Clin Invest Arterioesclerosis. 1998; 10: 65-73.
17. Malhotra A, Shafiq N, Arora A, Singh M, Kumar R, Malhotra S. Dietary interventions (plant sterols, stanols, omega-3 fatty acids, soy protein and dietary fibers) for familial hypercholesterolaemia. Cochrane Database Syst Rev. 2014; 10; 6:CD001918.
18. Braamskamp MJ, Hutten BA, Wiegman A. Early initiation of statin treatment in children with familial hypercholesterolaemia. Curr Opin Lipidol. 2015; 26: 236-9.
19. Vuorio A, Kuoppala J, Kovanen PT, et al. Statins for children with familial hypercholesterolemia. Cochrane Database Syst Rev. 2014; 23; 7:CD006401.
20. McCrindle BW. Familial hypercholesterolemia in children and adolescents. Curr Opin Lipidol. 2012; 23: 525-31.
21. Wiegman A, Gidding SS, Watts GF, et al; European Atherosclerosis Society Consensus Panel. Familial hypercholesterolaemia in children and adolescents: gaining decades of life by optimizing detection and treatment. Eur Heart J. 2015 doi: 10.1093/eurheartj/ehv157.
22. Mata P, Alonso R, Ruiz A, et al. Diagnóstico y Tratamiento de la Hipercolesterolemia Familiar en España. Documento de Consenso. Semergen. 2015; 41: 24-33.
23. Muñoz MT, Alonso M, Oyarzabal M, et al. Atorvastatin versus Colestipol in children and adolescents with familial hypercholesterolemia: safety and efficacy. Hrom Res. 2004; 62 (suppl 2): 196.
24. Gouni-Berthold I, Berthold HK. Familial hypercholesterolemia: etiology, diagnosis and new treatment options. Curr Pharm Des. 2014; 20: 6220-9.
25. Davidson M. The efficacy of colesevelam HCl in the treatment of heterozygous familial hypercholesterolemia in pediatric and adult patients. Clin Ther. 2013; 35: 1247-52.
26. Clauss S, Wai KM, Kavey RE, Kuehl K. Ezetimibe treatment of pediatric patients with hypercholesterolemia. J Pediatr. 2009; 154: 869-72.
27. Manlhiot C, Larsson P, Gurofsky RC, Smith RW, et al. Spectrum and management of hypertriglyceridemia among children in clinical practice. Pediatrics. 2009; 123: 458.
28. Cuchel M, Mehageer EA, du Toit Theron H, et al. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: A single-arm, open-label, phase 3 study. Lancet. 2012; 5: 40-6.
29. Stein EA, Gipe D, Bergeron J, et al. Effect of a monoclonal antibody to PCSK9, REGN727/SAR236553, to reduce low-density lipoprotein cholesterol in patients with heterozygous familial hypercholesterolaemia on stable statin dose with or without ezetimibe therapy: A Phase 2 randomised controlled trial. Lancet. 2012; 380: 29-36.
30. Page MM, Bell DA, Hooper AJ, Watts GF, Burnett JR. Lipoprotein apheresis and new therapies for severe familial hypercholesterolemia in adults and children. Best Pract Res Clin Endocrinol Metab. 2014; 28: 387-403.
| Caso clínico |
|
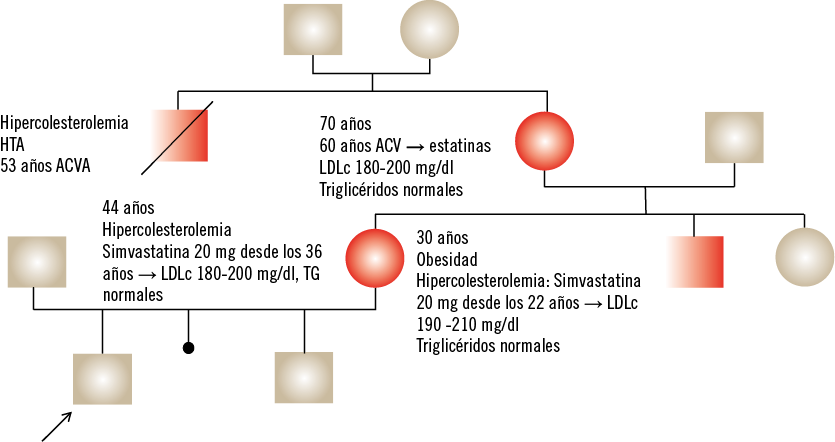
Varón de 10 años, derivado a nuestra consulta desde otro centro hospitalario, por hipercolesterolemia desde los dos años, en tratamiento con dieta, resincolesteramina y esteroles vegetales. Antecedentes personales: Embarazo y parto normal. PRN: 2.900 g (–1,5 DE), LRN: 48 cm (–1,6 DE). Sin incidencias en el periodo neonatal. Despistaje de enfermedades metabólicas: negativo. Alimentación normal, sin intolerancias. Desarrollo psicomotor normal. Antecedentes familiares: (ver Fig. 5).
Figura 5. Antecedentes familiares. Exploración física: Peso: 40 kg (+0,34 DE), Talla: 149 cm (+1,33 DE), IMC: 18 kg/m2 (+0,4 DE). TA: 105/60 mm HG (<P90). Buen estado general. Fenotipo armónico. Sin bocio. Sin estigmas cutáneos de dislipemia. Sin alteraciones cutáneas ni adenopatías significativas. ACP: normal. Abdomen: normal. Estadio I de Tanner (testes de 3 ml, pubarquia 1, axilarquia a). Estudios complementarios: • Hemograma y bioquímica general: normal. • Perfil tiroideo normal. • Vitaminas liposolubles normales. • Edad ósea: 10 años y 6 meses • Niveles de lipoproteínas desde el inicio de tratamiento a los 2 años (Tabla V). • Estudio genético del receptor LDL en sangre al paciente y a la madre: - Mutación clase A del receptor LDL (c.283 T>G), que da lugar a un cambio de aminoácidos (p.Cys95Gly).
|