 |
| El Rincón del Residente |
E. Cid París*, A. Losada Pajares*, M.E. Rubio Jiménez**, M.J. Alija Merillas**
*MIR de Pediatría. **Médicos Adjuntos. Servicio de Pediatría. Hospital Universitario de Guadalajara. Guadalajara
El Rincón del Residente es una apuesta arriesgada de Pediatría Integral. No hemos querido hacer una sección por residentes para residentes. Yendo más allá, hemos querido hacer una sección por residentes para todo aquel que pueda estar interesado. Tiene la intención de ser un espacio para publicaciones hechas por residentes sobre casos clínicos, imágenes y revisión bibliográfica. ¡Envíanos tu caso! Normas de publicación en www.sepeap.org Se presenta el caso de un neonato de 24 días de vida que acude a urgencias con estreñimiento, distensión abdominal e irritabilidad. Se plantean al lector preguntas sobre el diagnóstico y manejo del caso. |
Pediatr Integral 2013; XVII(7): 525-528
Caso clínico MIR. Haz tu diagnóstico. Neonato con distensión abdominal e irritabilidad
Caso clínico
Anamnesis
Neonato de 24 días que acude al Servicio de Urgencias derivado por su pediatra por distensión abdominal e irritabilidad tras las tomas de 48-72 horas de evolución. Afebril, apetito conservado sin rechazo de las tomas. Lactancia materna exclusiva. Regurgitación habitual tras las tomas desde la primera semana de vida, que parece haberse incrementado en la última semana. Meconiorrexis a los 7 días de vida, posteriormente realiza deposiciones de características normales, pero cantidad escasa, entre los 10 y 17 días de vida. A partir de ese momento sólo expulsa gases, pero “mancha” el pañal con deposiciones tras cada toma.
Antecedentes personales
Embarazo controlado con serologías normales. Parto inducido por preeclampsia a las 39 semanas de edad gestacional, finalizado por cesárea por riesgo de pérdida de bienestar fetal (bradicardia fetal). APGAR 9/9, no precisó reanimación. PRN: 3.010 g (p10-15). Pérdida máxima de peso a las 48 horas de vida con posterior ganancia ponderal. Periodo neonatal sin incidencias. Calendario vacunal completo para su edad. No alergias medicamentosas conocidas. No enfermedades ni intervenciones quirúrgicas.
Antecedentes familiares
Padres sanos, no consanguíneos, de origen español. No enfermedades infecto-contagiosas en la familia en el momento actual.
Exploración física
P: 3.150 g (p<3). Tª: 36,4ºC. FC: 123 lpm. FR: 28 rpm. SatO2: 100%.
Aceptable estado general, quejoso e irritable a la exploración, aunque calma en los brazos de la madre. Normocoloración de piel y mucosas. Bien hidratado y perfundido. No exantemas ni petequias, no aspecto séptico. Eupneico, sin signos de distrés respiratorio.
No adenopatías significativas. Orofaringe y otoscopia bilateral sin hallazgos.
Auscultación cardiaca con ruidos cardiacos rítmicos, sin soplos. Auscultación pulmonar con murmullo vesicular conservado, con buena entrada de aire bilateral y sin ruidos patológicos.
Abdomen con importante distensión, con aspecto tenso y brillante, con patrón vascular superficial, timpanizado, con borgborismo marcado y muy molesto a la palpación. Pulsos femorales positivos y simétricos.
Exploración neurológica: consciente y reactivo a la manipulación. Fontanela anterior normotensa. No signos de focalidad neurológica. Tono y reflejos adecuados para su edad.
Pruebas complementarias
Hemograma: leucocitos: 12.930/mm3 (4.560 neutrófilos/mm3, 5.670 linfocitos/mm3, 2.650 monocitos/mm3, 50 eosinófilos/mm3). Hemoglobina: 14 g/dl; hematocrito: 40,8%; VCM: 92,7 fl; HCM: 31,9 pg; CHCM: 34,4 g/dl; RDW: 15,4%; plaquetas: 494.100/mm3.
Bioquímica: glucosa: 78 mg/dl; creatinina: 0,2 mg/dl; urea: 16 mg/dl; sodio: 130 mmol/L; potasio: 3,9 mmol/L; GPT: 23 U/L; proteínas totales: 91,4 g/L.
Gasometría venosa: pH 7,41, pCO2 34, bicarbonato: 21,6 mmol/L. Exceso de base -2,3 mmol/L.
Radiografía de abdomen (Fig. 1): marcada distensión de asas con ausencia de gas a nivel distal.
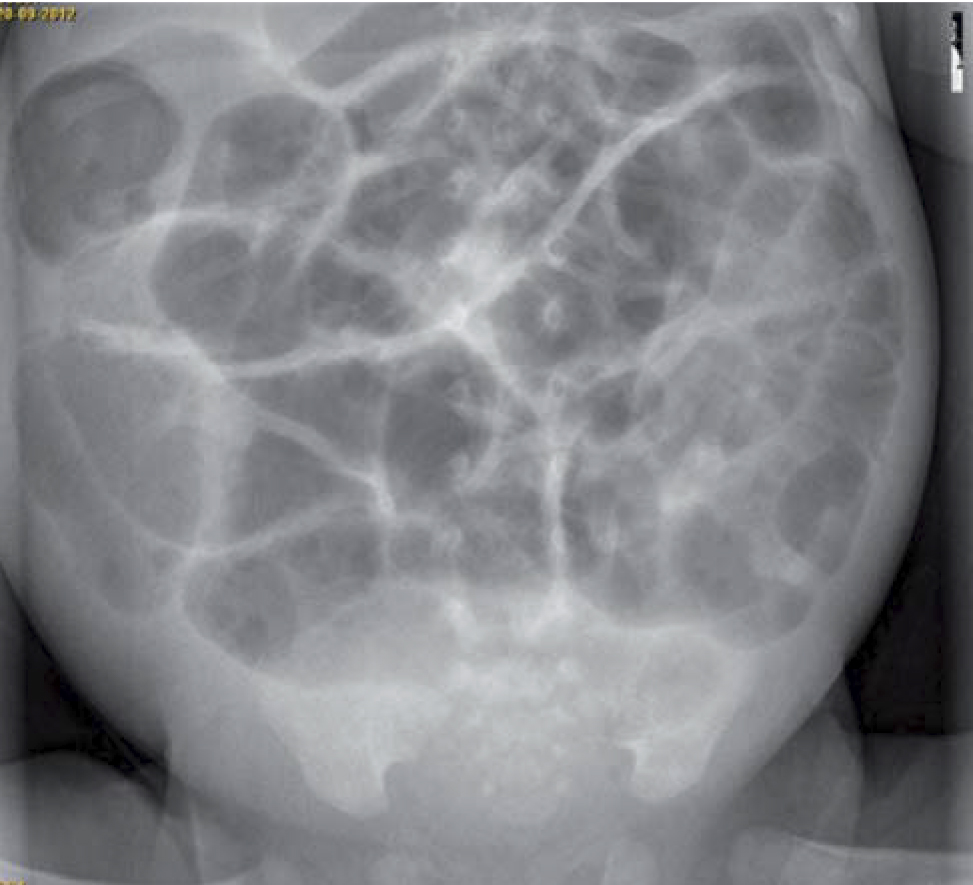 Figura 1. Radiografía de abdomen.
Figura 1. Radiografía de abdomen.
1. Ante la anamnesis, la exploración física y el resultado de las pruebas complementarias, ¿cuál de estos diagnósticos le parece más probable?
a. Síndrome de tapón meconial.
b. Enfermedad de Hirschsprung.
c. Íleo meconial.
d. Atresia intestinal.
e. Síndrome de colon izquierdo hipoplásico.
El retraso a la hora de realizar la primera deposición espontánea de meconio, asociado al cuadro de obstrucción intestinal funcional distal, con distensión abdominal marcada e irritabilidad, así como la radiografía de abdomen en la que se evidencia la ausencia de gas a nivel distal, deben hacernos sospechar de una enfermedad de Hirschsprung.
2. ¿Cuál es la prueba diagnóstica que da el diagnóstico de certeza en la patología que se sospecha?
a. Radiografía simple de abdomen.
b. Biopsia de la pared rectal.
c. TAC abdominal.
d. Manometría ano-rectal.
e. Enema opaco.
Respuesta correcta: b.
La radiografía de abdomen puede ofrecer signos de obstrucción intestinal distal, pero no permite el diagnóstico definitivo. El enema opaco ofrece más información, aunque su sensibilidad (70%) y especificidad (83%) continúan siendo inferiores a otras pruebas diagnósticas.
La manometría anorrectal tiene una alta sensibilidad (91%) y especificidad (94%), resultando muy útil en los casos de segmento agangliónico ultracorto, debido a la ausencia del reflejo anal inhibitorio característico de la enfermedad de Hirschsprung; sin embargo, la prueba diagnóstica de certeza es el estudio histológico mediante biopsia quirúrgica rectal que incluya la capa muscular, realizada mediante succión (sensibilidad del 93% y especificidad del 98%).
3. Una vez realizado el diagnóstico definitivo, ¿cuál es el tratamiento de esta patología?
a. Iniciar lactancia artificial con fórmula anti-estreñimiento.
b. Controlar el estreñimiento mediante el uso de laxantes osmóticos.
c. Realizar irrigaciones colónicas con SSF de forma crónica.
d. Irrigaciones colónicas con SSF y dilatación anal progresiva.
e. Una vez confirmado el diagnóstico, el tratamiento es siempre quirúrgico.
El tratamiento de la enfermedad de Hirschsprung es quirúrgico, con resección del segmento agangliónico y descenso del intestino sano al canal anal, preservando la función del esfínter interno. El diagnóstico precoz y el manejo de la obstrucción intestinal mediante nursing(sonda rectal intermitente, enemas de limpieza con suero salino fisiológico u otros) permiten resolver cuadros clínicos de suboclusión intestinal y enterocolitis, al mismo tiempo que mejora el cuadro clínico y consigue las condiciones adecuadas para la posterior intervención quirúrgica.
Evolución
Con la sospecha de enfermedad de Hirschsprung se decide ingreso con dieta absoluta, sueroterapia intravenosa y colocación de sonda orogástrica tipo replogle con aspiración continua, derivándose a un hospital terciario con Servicio de Cirugía Infantil.
Se inicia nutrición parenteral a través de un catéter epicutáneo central durante 5 días y se realizan irrigaciones colónicas con suero salino fisiológico diarias, que resultan efectivas, con expulsión de heces y gases. Se evidencia una mejoría progresiva del cuadro de distensión abdominal con desaparición de las regurgitaciones. Se realiza biopsia rectal por succión, que confirma el diagnóstico y, finalmente, se interviene quirúrgicamente sin incidencias.
Discusión
La enfermedad de Hirschspruhg (EH) es una malformación congénita que afecta a 1/5.000 recién nacidos vivos, con una relación 4:1 hombre/mujer, y que se caracteriza por la ausencia de la inervación intrínseca distal del intestino de longitud variable. Se localiza en la región recto-sigmoidea en el 75% de los casos, en el colon izquierdo y transverso en el 15% afecta a todo el colon en el 5% de los pacientes, al intestino delgado en el 3% y, por último, hay un número definido como de “segmento ultracorto” que se limita a los centímetros distales del canal anal y que afecta al 2% de los pacientes.
La expresión de este defecto es la ausencia de relajación involuntaria del esfínter anal interno al dilatarse el recto.
La causa es la colonización incompleta de las células derivadas de la cresta neural en su parte vagal. A diferencia de otras anomalías estructurales congénitas, esta enfermedad no presenta ninguna característica clínica que permita el diagnóstico en el periodo prenatal.
La EH ocurre de forma aislada en el 70% de los casos y la mayoría son formas de segmento corto. En el 30% aparece junto a otras anomalías congénitas: en un 12% existe una cromosomopatía (mayoritariamente, un síndrome de Down) y en el otro 18% habría otras alteraciones sindrómicas o distintas alteraciones aisladas.
En más del 90% de los casos, la clínica se inicia en el periodo neonatal, con el retraso en la expulsión del meconio mayor de 48 horas, en el caso del recién nacido a término y, posteriormente, con un cuadro de obstrucción intestinal funcional distal con ausencia de deposiciones, distensión abdominal, vómitos biliosos que se transforman en fecaloideos con el paso del tiempo. Un tacto rectal o una sonda rectal pueden permitir la salida explosiva de gas y/o meconio. Otras formas de presentación, mucho menos frecuentes, son en forma de enterocolitis en el periodo neonatal (9%), perforación intestinal o deshidratación severa.
El diagnóstico diferencial incluye descartar una obstrucción orgánica, el íleo meconial y el síndrome de colon izquierdo hipoplásico.
Respecto al diagnóstico, son importantes la radiografía simple, que revela asas de intestino dilatadas que pueden mostrar escasez de aire en recto, el enema de contraste, que presenta como hallazgo clásico un segmento de recto-sigma o colon distal estrecho y espástico con el colon proximal muy dilatado, y la manometría rectoanal, en la que, tras distender transitoriamente el canal anorrectal, no existe la relajación o reflejo rectoanal inhibitorio. Sin embargo, resulta esencial la realización de una biopsia rectal, que confirma el diagnóstico de EH al no encontrarse células ganglionares, con hipertrofia de troncos nerviosos e incremento inmunohistoquímico de acetilcolinesterasa.
Dicha biopsia debe realizarse, al menos, unos 3 centímetros por encima de la línea pectínea e incluir la mucosa y la submucosa.
El tratamiento de la EH, tras un cuidadoso manejo preoperatorio que incluye la realización de enemas de limpieza con suero salino fisiológico y sondaje rectal intermitente, es quirúrgico y está dirigido a colocar el intestino normal junto al ano, mediante el descenso de dicho intestino sano, tras resecar la zona aganglionar. Desde que, en el año 1948 se realizaron las primeras intervenciones con éxito (Swenson), se han desarrollado diversas técnicas que han permitido el descenso de la morbilidad y mortalidad y mejorar la calidad de vida.
Bibliografía
1. López-Alonso M, Losada Martínez A. Obstrucción intestinal en el periodo neonatal. En: Urgencias en Gatroenterología, Hepatología y Nutrición pediátrica. 1ª edición. Madrid: Ergon; 2011. p. 172-80.
2. Fernández Sánchez A, Barrena Delfa S, Salamanca Fresno L, Olivares Arnal P. Enfermedad de Hirschsprung y displasias intestinales. En: Tratado de Gastroenterología, hepatología y nutrición pediátrica aplicada de la SEGHNP. 1ª edición. Madrid: Ergon; 2011. p. 255-65.
3. Lee C, Lien R, Chian M, Yang PH, Chu SM, Fu JH, Lai JY. Clinical Impacts of Delayed Diagnosis of Hirschsprung’s Disease in Newborn Infants. Pediatr Neonatol. 2012; 53: 133-72.
4. De la Torre Mondragón. Enfermedad de Hirschsprung. Mitos y realidades a 120 años de su descripción. Acta Pediatr Mex. 2008; 29(3): 139-46.
5. De Manueles Jiménez J, De La Rubia Fernández L. Enfermedad de Hirschsprung. En: Protocolos de Gastroenterología, Hepatología y Nutrición. Protocolos de la AEP. 2ª edición. Madrid: Ergon; 2010. p. 47-52.
6. García C, Fantoral A. Caso clínico-radiológico para diagnóstico. Rev Chil Pediatr. 2002; 73(5): 500-3.
