 |
| Temas de FC |
S. Macías Franco, P. Rozas Reyes
Especialistas de Área en Oftalmología pediátrica. Hospital Universitario Central de Asturias. Oviedo
| Resumen
El conocimiento del genoma humano ha proporcionado en las últimas décadas mucha información sobre enfermedades con afectación ocular. Se trata de un tema en constante actualización y en auge debido a los desarrollos en Biología molecular y Terapia génica. El estudio de las bases genéticas en Oftalmología nos brinda diagnósticos nuevos cada día y nos proporciona las claves para tratar patologías incurables hasta ahora, que pueden acabar en ceguera. Se realiza una revisión sobre algunas de las más importantes y su manifestación en la infancia, con el objetivo de que el pediatra pueda sospechar, reconocer y actuar ante ellas. |
| Abstract
Over the last decades, the understanding of the human genome has provided a great deal of information on diseases with ocular involvement. This is a subject that is constantly being updated and is expanding due to developments in molecular biology and gene therapy. The study of the genetic basis in Ophthalmology provides new diagnoses every day and brings keys to treat pathologies incurable until now, which can ultimately lead to blindness. A review is made of some of the most important ones and their manifestations in childhood, so that the pediatrician can suspect, recognize and act upon them. |
Palabras clave: Alteraciones oculares genéticas; Distrofias retinianas; Terapia génica en Oftalmología; Catarata congénita; Neuropatías hereditarias.
Key words: Genetic ocular disorders; Retinal dystrophies; Gene therapy in Ophthalmology; Congenital cataract; Hereditary neuropathies.
Pediatr Integral 2023; XXVII (1): 7 – 15
OBJETIVOS
• Identificar patologías oculares de base genética y ciertas asociaciones.
• Poder realizar una anamnesis y exploración detallada y protocolizada en niños con sospecha de baja visión.
• Aprender de modo práctico ciertos signos y síntomas como el nistagmo, la fotofobia o la nictalopía, para poder orientar la búsqueda de enfermedades oculares en niños.
• Reconocer cuándo podría interesar realizar pruebas neurofisiológicas y/o estudios genéticos.
Patología ocular de base genética
Introducción
Hoy en día, se conocen en torno a 900 enfermedades hereditarias con afectación ocular, por lo que es lógico la aparición de subespecialistas en Oftalmología genética.
Es un hecho que el campo de la genética ha dado grandes pasos en un tiempo muy corto. En tan solo 30 años hemos pasado de hablar de “enfermedades raras”, donde la descripción de ciertas patologías era puramente clínica y/o fisiológica a la etapa actual, la era de la biología molecular, donde ya se busca tratar las enfermedades desde sus alteraciones genético-moleculares.
El objetivo de la siguiente revisión va a ser el de poder identificar ciertas patologías oculares de base genética, como médicos pediatras y oftalmólogos. Resultaría imposible comentar cada enfermedad con base genética, pues diariamente encontramos en la literatura reportes sobre asociación de gen-enfermedad, no solo enfermedades sindrómicas o pediátricas, sino que encontramos genética hasta en enfermedades seniles como la degeneración macular asociada a la edad.
A continuación, intentaremos aclarar por apartados ciertos conceptos para poder entender de manera teórica y práctica lo que nos concierne en este artículo.
El genoma humano
30 años para describir, comprender y editar el genoma humano.
En 1990 comienza el proyecto genoma humano que finaliza en 2003: su objetivo era el de determinar la secuencia de pares de bases que componen el ADN y mapear así todos los genes del genoma humano. No se conoce la función de todos ni por qué unos se expresarán fenotípicamente y otros no, pero este proyecto nos da el código y el avance, que desde ahí en genética ha sido creciente(1).
Actualmente, encontramos en la era de la Biología molecular donde se estudian los mecanismos genético-moleculares como base de las enfermedades para así no solo conocerlas o definirlas, sino tratarlas y curarlas. Sí, hablamos de lo que se conoce como Terapia génica. Hasta hace poco, “editar” el genoma humano suponía muchas limitaciones, por lo que parecía prácticamente imposible.
En Oftalmología disponemos del primer fármaco génico aprobado para amaurosis congénita de Leber tipo 2 (ACL), debida a mutaciones en el gen RPE65. El tratamiento se realiza mediante la inyección subretiniana de Voretigene Nepavenec (Luxturna®) a través de un virus vector (Adeno-associated viral vector serotype 2 o AAV2) que infecta la célula consiguiendo reemplazar el gen mutado por el gen sano. Este tratamiento ha conseguido que pacientes con ceguera puedan recuperar visión(2).
Se avecina un futuro prometedor. Las doctoras Charpentier y Doudna estudiaron el funcionamiento del sistema CRISPR-Cas9, mecanismo de defensa de Streptococcus Pyogenes frente a virus conocido como “tijeras moleculares”. En 2012 publicaron un estudio que proponía el uso de CRISPR-Cas9 como herramienta para la edición del genoma, consiguiendo reprogramar el sistema para reconocer y cortar fragmentos de ADN eliminando genes específicos, pero también modificar esos genes introduciendo la secuencia correcta(3). Dicho de otro modo, sustituir un gen patológico por uno sano. En el año 2020 recibieron el Premio Nobel de Química por su trabajo.
Cuándo y qué pruebas genéticas solicitar a un paciente
La anamnesis y la exploración clínica deben ser muy detalladas, del paciente y de todos los familiares posibles. Conocer datos pasados, nos brindará una excelente ayuda a la hora de saber por dónde empezar.
Ante la sospecha según los hallazgos clínicos de una enfermedad ocular hereditaria, el pediatra y el oftalmólogo deberán valorar la necesidad o utilidad de realizar pruebas genéticas. Para ello, deben contar con la formación específica o consultar a otro médico con experiencia en seleccionar e interpretar la prueba a realizar, así como poder proporcionar un correcto asesoramiento ante un resultado positivo.
Además, en Internet disponemos de múltiples recursos, bases de datos, bibliografía e incluso registros de profesionales. Entre otras, la web del Instituto Nacional de Investigación del Genoma Humano de Estados Unidos (NHGRI) dispone de gran cantidad de recursos didácticos para profesionales de la salud y población general(3-4).
Del mismo modo, no debemos dejar de lado resultados denominados “mutaciones de significado incierto” y es recomendable repetir los estudios genéticos tras 2 años ante estos resultados. Como hemos visto previamente, cada vez se conoce más sobre el genoma y las alteraciones sin explicación, que en un futuro pueden tenerla. Enfermedades que hoy no tienen cura podrían tenerla.
En la consulta del pediatra
El pediatra será el enlace entre el paciente y el oftalmólogo. Es de suma importancia realizar una historia detallada y una exploración protocolizada del niño.
Anamnesis
• Árbol genealógico: hasta donde podamos alcanzar. Es importante preguntar y anotar consanguineidad directa o sospecha. En ambientes rurales o ciertas etnias es más frecuente el parentesco y la aparición de enfermedades hereditarias recesivas.
• Antecedentes familiares: de inicio insistir sobre los antecedentes familiares con algún tipo de defecto visual o cirugías oftalmológicas previas. Posteriormente y tras la exploración, es recomendable volver al árbol genealógico para anotar antecedentes de otro tipo que puedan estar en relación con las patologías que sospechemos. También nos puede ayudar solicitar fotografías familiares.
– Ante un niño con una gran miopía axial o antecedentes de desprendimiento de retina, vamos a buscar signos, síntomas en él y en su familia de laxitud articular, pues podemos encontrar una colagenopatía.
– En el caso de las fotografías, podemos encontrar rasgos fenotípicos muy interesantes. Llegamos a ver con más frecuencia de lo esperado, familias completas con ptosis bilateral sugestiva de enfermedades mitocondriales sin estudiar.
• Antecedentes personales: desarrollo motor, cognitivo y conductual del paciente desde el nacimiento.
– Preguntar sobre el comportamiento referente a la atención del niño ante imágenes y objetos y sobre la destreza a la hora de realizar actividades donde la visión es fundamental.
– La habilidad para moverse. Una retracción de campo visual puede hacernos ver a un niño “torpe” a la hora de bajar las escaleras, pese a que al conservar la visión intacta central nos esté señalando un avión en el cielo. El paciente con baja visión en casa suele estar más cómodo, pues tiene aprendida las distancias y la localización del mobiliario y objetos. En lugares desconocidos se mostrará más inseguro.
– Es muy importante poder conocer cómo actúa el niño en ambientes con distinta luz. La fotofobia suele ser referida por los padres, pero el miedo a la oscuridad no, y no conocer esto puede llevarnos a solicitar pruebas innecesarias y no realizar las pertinentes. Por ejemplo, realizar una resonancia magnética con sedación, en un niño con baja visión con una distrofia de conos no manifiesta en la exploración de fondo de ojo, por ser aún un estadio precoz, en lugar de realizar pruebas neurofisiológicas y genéticas que nos darían el diagnóstico.
• Enfermedades y cirugías previas: lo obvio y lo que aparentemente no tiene relación:
– La trisomía 21 se asocia a múltiples patologías oftalmológicas (miopía, estrabismo, cataratas), pero el paciente viene diagnosticado desde el día que nace (la mayoría ya con la genética realizada prenatalmente), por lo que solo tenemos que diagnosticar las manifestaciones oculares.
– Manchas café con leche y neurofibromatosis tipo 1, donde varios de los criterios diagnósticos son oftalmológicos (nódulos de Lisch, neurofibromas palpebrales y orbitarios, glioma en vía visual, hipoplasia esfenoidal).
– Hay que profundizar en casos de casualidad y buscar causalidad: ¿Polidactilia + miedo a la oscuridad? Busquemos desde el principio afectación renal y problemas neuroendocrinos, pues podríamos estar ante un síndrome de Bardet-Bieldt. No existe tratamiento para la distrofia de conos y bastones, que en casos progresivos puede resultar en una ceguera, pero si desde niño se le deriva a un centro especializado en baja visión podremos ayudarlo a obtener una mejor calidad de vida.
– Patologías donde el ojo será el único afectado: nistagmo + fotofobia → albinismo ocular.
Exploración oftalmológica (Fig. 1)

Figura 1. A. Exploración. Toma de agudeza visual. B. Test de Snellen y Test de Ishihara. C. Hirchsberg. D. Leucocoria.
• Agudeza visual: dependiendo de la edad se usarán distintos test (mirada preferencial, Snellen, Piggassou, letras. También se valorará visión lejana y cercana.
En una consulta de primaria, en niños muy pequeños, solo podemos saber si fija y sigue el objeto que le estamos presentando. La intolerancia ante la oclusión de un ojo nos puede indicar baja visión del ojo que está libre.
• Test de Brückner: con oftalmoscopio directo, valorar el reflejo rojo retiniano. Muy importante si existe leucocoria (reflejo blanco) y remitirlo de manera urgente a un oftalmólogo para descartar la presencia de retinoblastoma, tumor maligno ocular más frecuente en niños.
• Motilidad intrínseca: tamaño de pupila en la luz y oscuridad.
– Asimetría: pupila miótica (Horner, sinequias). Pupila midriática (Adie, disgenesias del segmento anterior como la aniridia, DPAR).
– DPAR (defecto pupilar aferente relativo): pupilas midriátricas que no reaccionan a la luz, nos dan información sobre daños en vía visual y sospecha de muy baja visión o ceguera.
• El test de Hirchsberg: consiste en valorar el reflejo de la luz de una linterna en las pupilas, con el niño mirando de frente. Si no se presenta en el centro de ambas, existe una desviación (estrabismo). Un motivo frecuente de consulta es el epicanto, confundido con un estrabismo convergente, por ello familiarizarse con la realización sistemática del test de Hirchsberg es importante.
• Motilidad extrínseca: movimientos oculares. Hacer posteriormente seguir la luz, una imagen o incluso ya un sonido para descartar limitaciones de movimientos oculares en todas las posiciones de la mirada con ambos ojos (versiones) y por separado (ducciones).
• Nistagmo: la presencia de nistagmo desde muy temprana edad es signo de baja visión.
• Reflejos: sacádicos, de seguimiento lento, vestibular, optocinético. Informan sobre el estado de la vía visual posterior y daño cerebral.
• Transparencia de medios: opacidades corneales no nos permiten ver con claridad iris y pupila. En opacidades en cristalino (catarata) veremos reflejo rojo pobre o inexistente.
• Anomalías faciales y palpebrales: ptosis, epicanto, telecanto, epiblefaron, alteraciones en pestañas, puntos lagrimales, lesiones o manchas en la piel, disposición ósea orbitaria. Palpar y descartar la presencia de masas.
• Campimetría por confrontación: en niños colaboradores, de forma monocular y con la fijación al frente, presentamos imágenes en distintos puntos del campo visual. Ante una reducción concéntrica, el niño no verá las imágenes cuando no las presentamos en el centro.
• Test de Ishihara: hoy en día, hay aplicaciones para dispositivos electrónicos que lo disponen de manera gratuita. Es lo que comúnmente se conoce como “test de colores” (aunque no el único), muestra unas láminas con números donde dependiendo de la afectación se verán o no. En niños que no conocen los números, pero colaboradores, se lo proponemos como un juego donde con un dedo le pedimos que siga el “camino” que marca el mismo. Útil en el diagnóstico de discromatopsias. Lo más frecuente es el daltonismo (y que se presente en varones en varias generaciones de una familia), pero un resultado anómalo puede indicar daño macular o en el nervio óptico.
Enfermedades oculares de base genética
Catarata
Primera causa de ceguera reversible en el mundo, pero su aparición en niños puede traer daños visuales irreparables y resultar en una baja visión si no se actúa precozmente.
En adultos se ha reconocido como un fenómeno multifactorial que implica relaciones entre genética y factores ambientales.
En niños es más deducible que exista un origen genético o en el desarrollo embrionario. Las cataratas congénitas e infantiles pueden aparecer de forma aislada o asociadas a otros trastornos sistémicos. Se han encontrado más de 100 genes relacionados con cataratas sindrómicas, pero también se están viendo mutaciones genéticas donde la catarata es la única manifestación clínica.
Se estima que aparece como un trastorno mendeliano en 1/10.000 nacimientos y pueden desarrollarse en cualquier momento de la vida, aunque la mayoría son congénitas, pero con distinto fenotipo. La forma más común de herencia es la autosómica dominante (casi un 50 %), seguido del autosómico recesivo (18 %). Aproximadamente, un 30 % son esporádicas sin antecedentes familiares claros(5).
En cuanto al fenotipo, la mayoría son nucleares seguidas de polares posteriores (Fig. 2).

Figura 2. A. Catarata congénita. B. Cirugía de catarata congénita.
Cat-Map, se trata de un mapa de cromosomas online y una base de datos de libre acceso centrada en los genes y la descripción clínica de las cataratas registradas en ella, así como asociaciones sindrómicas(6).
Glaucoma congénito primario
Epífora, fotofobia y blefarospasmo.
Tipo de glaucoma infantil que se desarrolla antes o después del nacimiento, dependiendo del grado de afectación del ángulo iridocorneal. La afectación de las estructuras oculares del drenaje del humor acuoso que se encuentran en dicha zona ocasionará un aumento de la presión ocular. Esto provocará que en niños se desarrolle buftalmos (diámetro corneal aumentado por la distensión gracias a la plasticidad corneal prenatal y en los primeros 2 años de vida) y una miopía (crecimiento axial por elasticidad escleral del mismo modo). Los pacientes más graves pueden nacer con la córnea de color azulado, debido a que ese aumento de presión sobrepasa la capacidad del endotelio corneal y desgarra estructuras importantes en la transparencia de la misma.
La presión mantenida daña el nervio óptico irreversiblemente provocando importante discapacidad visual si esa situación es avanzada y si no se revierte pronto.
En formas menos evidentes, sin buftalmos claros y transparencia de medios, la tríada clínica de: epífora, fotofobia y blefarospasmo, debe poner en alerta y ser reconocida por el pediatra.
En cuanto al patrón de herencia es complejo, la mayoría aparece de manera esporádica, pero podemos encontrar asociación familiar hasta en un 40 % de los casos, donde se propone un patrón autosómico recesivo, donde se ha descrito especialmente relación con alteraciones en el gen CYP1B(7).
Enfermedades hereditarias de la retina
Grupo amplio de enfermedades, generalmente progresivas y con consecuencias devastadoras en la visión, acabando en ceguera legal en la mayoría.
Presentan baja prevalencia y se consideran y cumplen criterios de enfermedades raras. Pese a ello, es importante reconocerlas y estudiarlas, no solo por poder realizar el consejo genético, sino porque en la actualidad hay ensayos clínicos con tratamientos prometedores en el ámbito de la terapia génica. A continuación expondremos algunas de ellas.
Retinosis pigmentaria (RP)
Distrofia retiniana hereditaria más prevalente (1:4.000 personas) y con mayor número de genes y loci asociados (>90), los primeros descritos en la década de los 80(8). Forma parte hasta en un 20 % de los casos de una enfermedad sindrómica y estos casos suelen ser diagnosticados durante la infancia y adolescencia (Tabla I).
El daño se origina en los bastones, pero progresivamente acaba afectando a los conos y al resto de células retinianas. Hay mucha variabilidad en cuanto a la edad de manifestación de los primeros síntomas y la velocidad de progresión y afectación de la visión.
La enfermedad inicialmente cursa con nictalopía (baja visión en oscuridad) y posterior reducción concéntrica del campo visual, bilateral y simétrica, llegando en muchos casos a la ceguera. En la exploración del fondo de ojo, lo más típico es la tríada clásica: palidez del nervio óptico + atenuación vascular + espículas periféricas (Fig. 3). Asocia cataratas y, en estadios finales, edema macular que precipitará la pérdida de visión central.
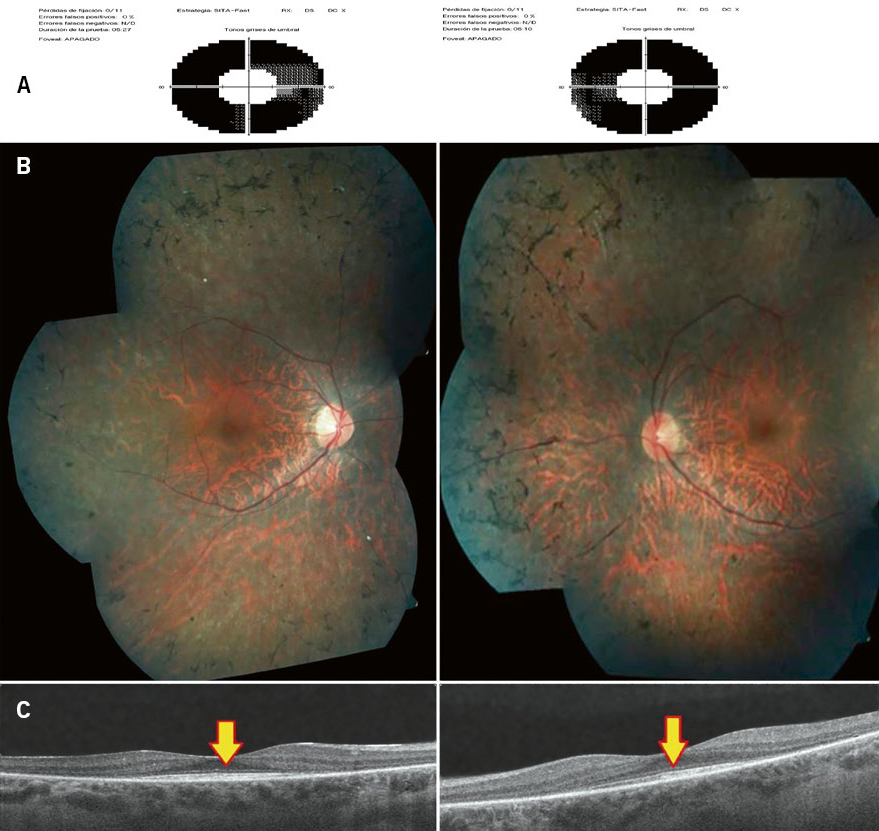
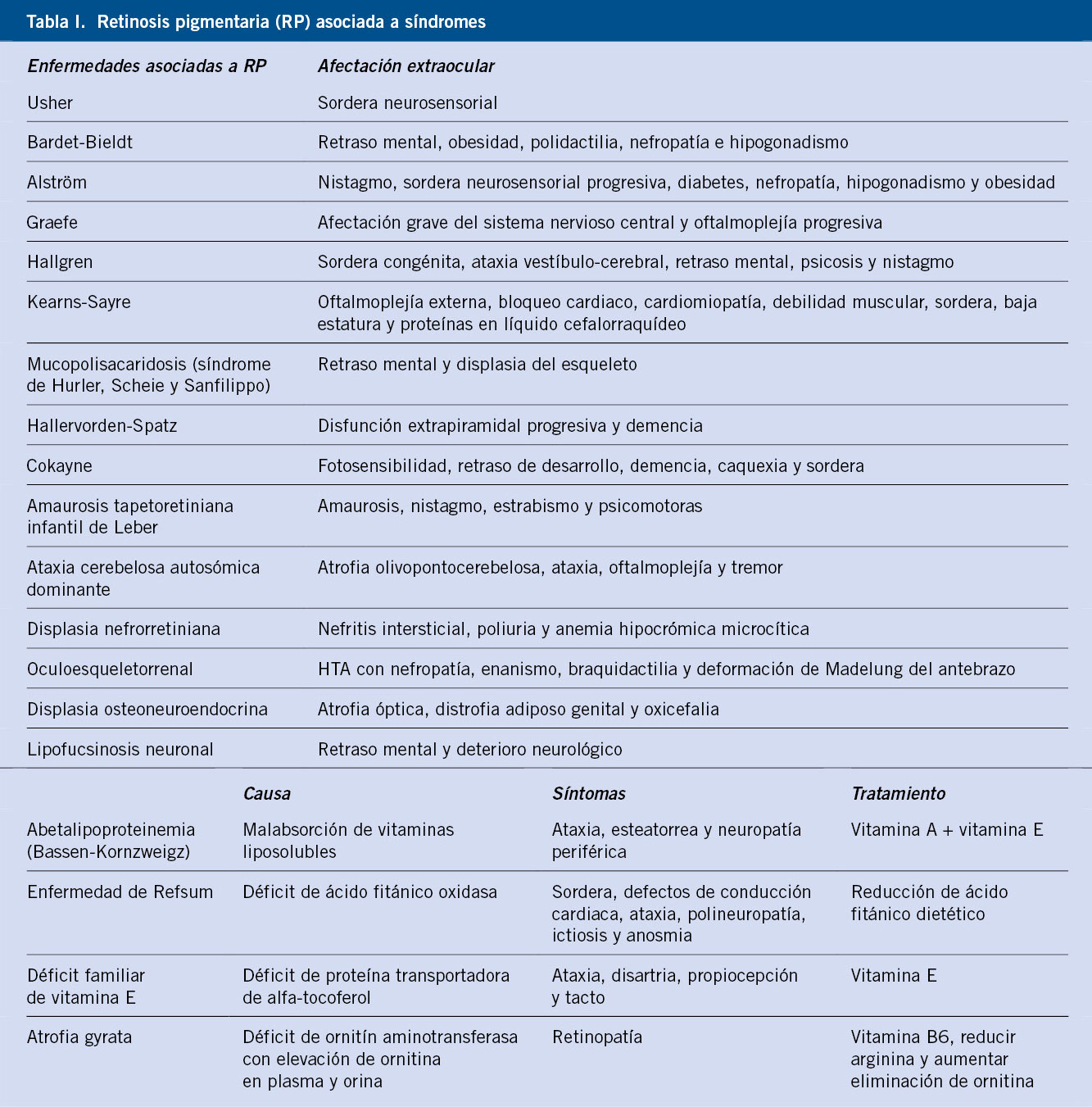
Figura 3. A. Retinosis pigmentaria. Retracción concéntrica de campo visual bilateral. B. Composición de fondo de ojo derecho e izquierdo. C. Imágenes de tomografía de coherencia óptica que muestran preservación de capas externas en retina central.
Distrofia progresiva de conos y distrofia de conos y bastones (CORD)
Menos prevalencia (1:20.000/100.000) que la RP, estas suelen aparecer desde edades tempranas con una gran fotofobia, afectación en la visión de colores y pérdida de visión central por afectación inicial de los conos. El diagnóstico puede suponer un reto, pues en muchos casos podríamos creer que estamos ante un paciente pediátrico simulador, ya que los cambios en el fondo de ojo (maculopatía en ojo de buey) (Fig. 4) no suelen estar presentes al principio. La electrofisiología puede afectarse desde estadios precoces, por lo que será muy útil en la orientación del paciente con esta clínica.
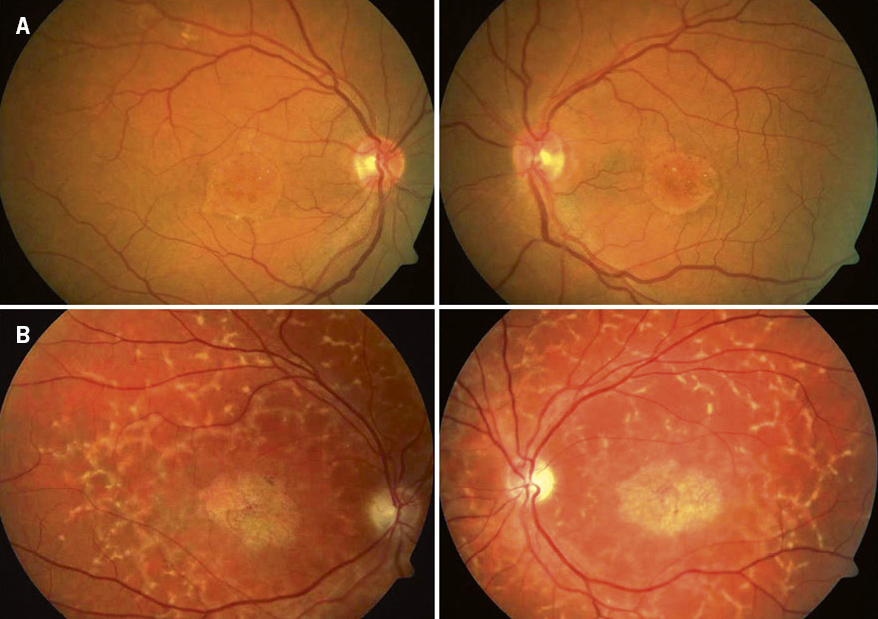
Figura 4. A. Distrofia hereditaria de retina. Distrofia de conos. B. Enfermedad de Stardgardt fundus flavimaculatus.
Amaurosis congénita de Leber (ACL)
Se trata de un conjunto de distrofias muy severas, donde se afectan conos y bastones de forma muy precoz, de herencia recesiva, con más de 20 genes descritos en relación(9). A pesar de su baja prevalencia (1:33.000/80.000), es de vital importancia conocerla, pues los pacientes con una mutación concreta en el gen RPE65 pueden recibir tratamiento con la primera terapia génica aprobada con Voretigene neparvovec (Luxturna®) en 2018. Para ello, al menos, tienen que presentar células retinianas viables, por lo que debe detectarse en la infancia(10).
El inicio es muy precoz, niños pequeños que presentan una agudeza visual menor de <1/20, nistagmo e hipermetropía alta. Un signo a conocer por el pediatra y el oftalmólogo es el signo óculo-digital, donde el niño se aprieta y frota los ojos; pero lo descrito no es patognomónico ni una tríada clásica, sino que es común en enfermedades de inicio precoz con muy baja visión, por lo que habrá que descartar otras patologías y realizar un estudio electrofisiológico previamente. Hemos de decir que la experiencia nos ha demostrado que, en muchos casos, por la colaboración y la baja visión del niño no es posible realizarlos, por lo que en estos casos concretos, si no obtenemos otro diagnóstico mediante la exploración, es cuando creemos que no hay que esperar para realizar un estudio genético. Aunque encontrar el gen concreto sea dentro de una enfermedad rara, algo todavía más raro.
Enfermedad de Stardgardt
Se trata de una distrofia macular y la segunda distrofia hereditaria de retina (DHR) más frecuente (1:8.000/10.000).
A diferencia de las anteriores, las distrofias maculares afectan al área central de la retina, la mácula. Tendremos, por tanto, una pérdida de campo central/paracentral, lo que lleva a grandes limitaciones en la agudeza visual y, sobre todo, en la lectura y de cerca, aunque existe una amplia variabilidad en cuanto al comienzo de la enfermedad y severidad. El gen afectado con mayor frecuencia es el ABCA4 y su herencia es autosómica recesiva. Este gen puede verse afectado en casos de RP y CORD. Su alteración produce un aumento de lipofucsina en la retina externa con degeneración de los fotorreceptores (Fig. 4). Se inicia en muchos casos en la pubertad-adolescencia y puede confundirse con la distrofia de conos por su afectación central, ausencia de signos en el fondo de ojo precoces, aunque a diferencia de la anterior, las visiones suelen ser mejores, las pruebas neurofisiológicas tardan más en alterarse y la fotofobia no es tan intensa.
Las gafas con filtros de absorción selectiva son muy útiles y hay ensayos sobre medicamentos; evitar el acúmulo de la lipofuscina e incluso sobre trasplante retiniano.
En estos pacientes se desaconseja tomar vitamina A, pues se considera uno de los responsables etiopatogénicos.
Ceguera nocturna estacionaria congénita
Es debida a un fallo en la fototransducción (transmisión de información entre fotorreceptores y células bipolares). Cursa con nictalopía en la infancia y visiones relativamente buenas, miopía y fondo de ojo normal estables en el tiempo, lo que puede llevar a infradiagnosticarla. El electrorretinograma será lo que nos dé el diagnóstico y el puente hacia realizar un estudio genético(11).
Daltonismo y disfunciones de conos no progresivas
El daltonismo es una condición en la cual el paciente no puede distinguir ciertos colores. Es hereditario con herencia ligada a X, por lo que se da más frecuentemente en varones; de hecho, según algunas series, hasta un 8 % de los hombres pueden padecer algún tipo de alteración en la percepción de los colores.
Una discromatopsia adquirida nos debe llevar a sospechar enfermedades del nervio óptico o retina, por lo que si nunca se sospechó de daltonismo congénito habrá que descartar estas.
Característicamente, hay una reducción de los conos en la retina. La forma más severa es la acromatopsia congénita completa, enfermedad AR con múltiples genes descritos asociados. La visión se presenta muy baja desde el nacimiento con fotofobia importante, nistagmo, escotoma central y ceguera completa de colores. Los monocromatismos de conos hacen referencia a la falta de 2 de los 3 tipos de conos. Esto produce incapacidad en la distinción de los colores(12).
Aunque actualmente existen ayudas ópticas, por lo general en el daltonismo (salvo en la acromatopsia) no se ven limitaciones en la vida del paciente, por lo que debemos tranquilizar a los padres en la consulta, no alentarlos a forzar al niño a juegos donde la discriminación de colores sea importante y, sobre todo, normalizar esta situación tanto en casa como en el colegio.
Enfermedades hereditarias del nervio óptico (Fig. 5)
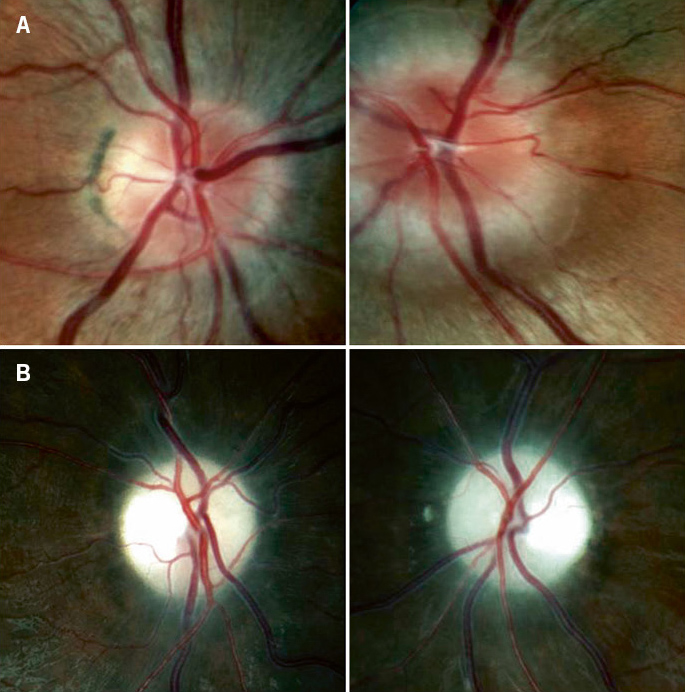
Figura 5. A. Edema de papila asimétrico en la neuropatía óptica hereditaria de Leber (NOHL). B. Atrofia óptica.
Enfermedades donde existe una afectación bilateral del nervio óptico.
Los síntomas de las neuropatías incluyen: pérdida de agudeza visual y alteración en la percepción de colores y sensibilidad al contraste. Los defectos en el campo visual son centrales y, al igual que en otras enfermedades con grave afectación visual precoz, podremos encontrar nistagmo.
Neuropatía óptica hereditaria de Leber (NOHL o LHON)
Su prevalencia es baja 1:45.000. El 90 % se manifiesta entre la segunda y tercera década de vida. En cuanto a su etiología, se trata de la enfermedad más común causada por mutaciones del ADN mitocondrial, por tanto, de herencia materna. Es de fácil estudio genético, pues el 90-95 % está causada por 3 mutaciones (mutaciones primarias). Se cree que no es suficiente presentar la alteración para el desarrollo de la enfermedad, sino que los factores ambientales como el tabaco, alcohol o déficit vitamínicos, influyen considerablemente en ello.
Suele comenzar con una fase presintomática que puede pasar desapercibida, donde solo se afecta la percepción de colores y sensibilidad al contraste. Posteriormente, se produce una disminución de agudeza visual bilateral, pero que puede ser de inicio diferido de hasta 6-8 semanas en el ojo contralateral. Se trata de una disminución importante con agudez visual de 0,1 o menos, indolora (a diferencia de las neuritis) y sin DPAR (defecto pupilar aferente relativo). En la exploración de fondo de ojo se puede ver hiperemia y tortuosidad vascular, pero puede ser imperceptible y normal en hasta el 20 % de los pacientes.
Finalmente, la fase crónica se define por la atrofia en la capa de fibras del nervio óptico unos 6 meses tras el inicio. Algunos pacientes pueden recuperar espontáneamente la visión incluso años después.
Desde el año 2015 existe un fármaco aprobado, Idebenona (Raxone®). Su uso está reservado para pacientes que lleven menos de 12 meses desde el inicio de los síntomas. Por tanto, la sospecha clínica nos debe hacer actuar rápido(13).
Atrofia óptica dominante (AOD o enfermedad de Kjer)
De herencia autosómica dominante, se estima una prevalencia de 1:50.000, pero con penetrancia y expresividad variables. Se debe a una mutación en el cromosoma 3, gen OPA1 en la mayoría de los casos.
Comienza antes de la primera década de vida, con pérdida de agudeza visual progresiva y bilateral variable, pero que en un 40 % llegarán a cumplir criterios de ceguera legal. Presentan defectos de visión de colores. A diferencia de la LHON, no se ve mejoría espontánea y, al igual que esta, se debe insistir en la abstención de tóxicos (alcohol, tabaco y déficits nutricionales).
Miopía
La miopía degenerativa supone una de las principales causas de discapacidad visual irreversible.
Merece un capítulo aparte, ya que es la patología ocular más prevalente en el mundo occidental, lo que supone un problema de salud pública con un gran impacto socioeconómico.
En 2016, Holden et al. publicaron un metaanálisis donde incluyeron 145 estudios publicados desde 1995 y más de 2 millones de pacientes. El objetivo fue describir la prevalencia de la enfermedad y lo más interesante, realizar una estimación futura de la misma. Según la tendencia, de 163 millones de miopes en el año 2000 (2,7 % de la población global), pasaríamos a 517 millones (6,1 %) en el 2030 y 938 millones de miopes (casi un 10 % de la población mundial) en el 2050(14). Si ya estos datos nos podrían parecer alarmantes, si se analiza y revisa la literatura actual y separamos por grupos de edad y poblaciones, aún es más llamativo. En ciertos núcleos urbanos de Asia del este, ya la prevalencia ha alcanzado entre el 80-90 % de los jóvenes de entre 17-18 años(15). En Europa ha habido también un crecimiento exponencial con un 46 % de los jóvenes de 25 años afectados de miopía.
Podemos hablar de que hay una verdadera epidemia miópica. En cuanto a la enfermedad, partimos de una base genética compleja y en la que los factores ambientales juegan un papel importante; de hecho, muchas de las estrategias actuales en cuanto al control de la progresión de la miopía en el niño trata de controlar esos factores.
Los estudios coinciden en que las actividades al aire libre prolongadas reducen la aparición y progresión de la miopía. Así como, vivir en núcleos urbanos y el alto nivel educativo se pueden considerar de riesgo, mientras que el trabajo de cerca o el uso de pantallas no han demostrado ser un factor de riesgo independiente.
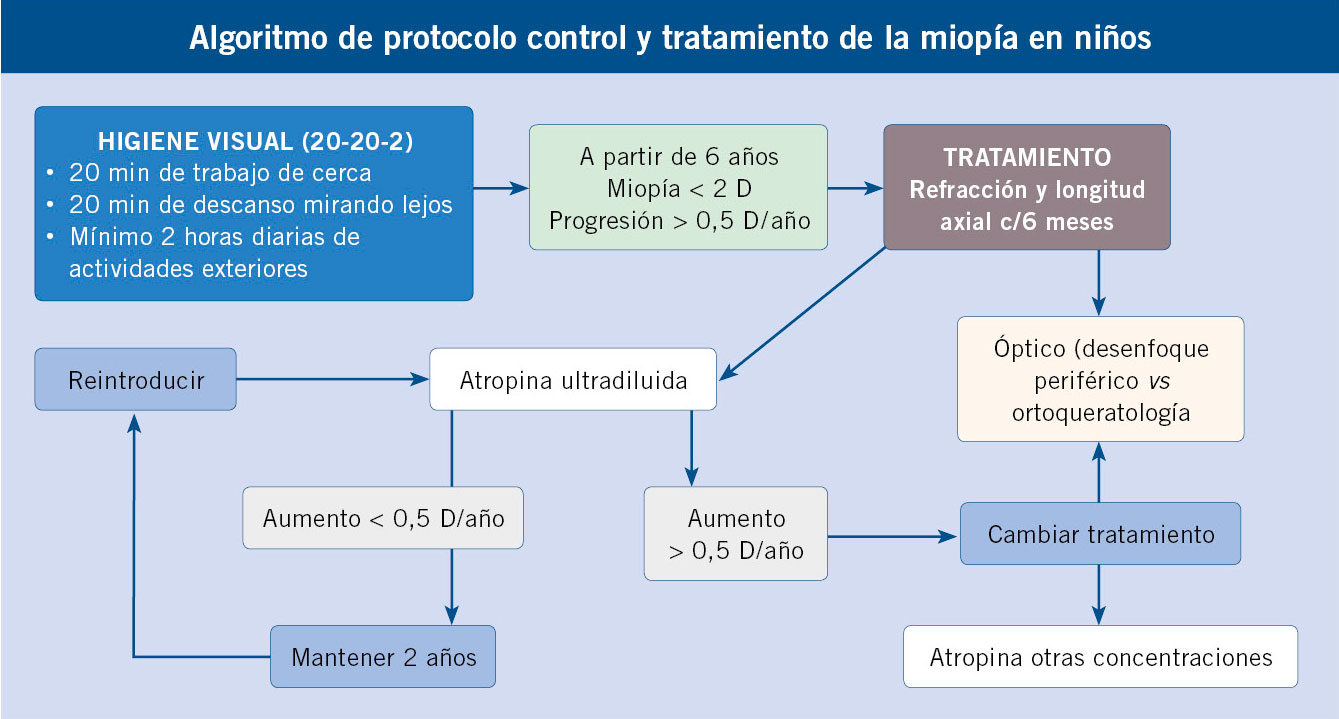
En cuanto al tratamiento, disponemos de dos opciones: tratamiento óptico, bien con lentes de desenfoque periférico o la ortoqueratología y el tratamiento médico con atropina ultradiluida, cuyo mecanismo exacto de actuación no se conoce, pero ha demostrado ser el tratamiento más eficaz para el control de la progresión de miopía(16). Aún, en España, no existe comercializado y debe recetarse como fórmula magistral, siendo financiado por la mayoría de sistemas sanitarios en nuestro país. Actualmente, pautamos 1 gota de atropina al 0,01 % por la noche en cada ojo en niños miopes con progresión de miopía de entre 6-12 años durante dos años. Se trata de un tratamiento muy bien tolerado y seguro. En cuanto a miopías magnas (mayores a 6 dioptrías), no hay estudios aún que evalúen su eficacia.
Función del pediatra en Atención Primaria
• Realizar una anamnesis detallada sobre los antecedentes personales y familiares del niño.
• Exploración completa del niño con sospecha de déficit visual.
• Orientar la exploración desde la consulta de primaria en búsqueda de patologías de base genética. Conocer los signos y síntomas de las enfermedades del segmento anterior (cataratas, glaucoma), posterior (retina y nervio óptico) y defectos refractivos (miopía).
• Conocer los criterios de derivación al especialista en Oftalmología infantil y reconocer el carácter preferente de patologías genéticas que podrían beneficiarse de tratamiento, así como de un estudio genético.
Conflicto de intereses
No hay conflicto de interés en la elaboración del manuscrito. Declaración de intereses: ninguno.
Bibliografía
Los asteriscos reflejan el interés del artículo a juicio de las autoras.
1. Nurk S, Koren S, Rhie A, Rautiainen M, Bzikadze AV, Mikheenko A, et al. The complete sequence of a human genome. Science. 2022; 376: 44-53. DOI: 10.1126/science.abj6987.
2.** Wang X, Yu C, Tzekov RT, Zhu Y, Li W. The effect of human gene therapy for RPE65-associated Leber’s congenital amaurosis on visual function: a systematic review and meta-analysis. Orphanet J Rare Dis. 2020; 15: 49. DOI: 10.1186/s13023-020-1304-1.
3.*** Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014; 346: 1258096. DOI: 10.1126/science.1258096.
4.** Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, et al. GeneReviews®. Seattle (WA): University of Washington, Seattle; 1993-2022. What’s New in GeneReviews. Disponible en: https://www.ncbi.nlm.nih.gov/books/NBK132722/.
5. Berry V, Georgiou M, Fujinami K, Quinlan R, Moore A, Michaelides M. Inherited cataracts: molecular genetics, clinical features, disease mechanisms and novel therapeutic approaches. British Journal of Ophthalmology. 2020; 104: 1331-7.
6.** Shiels A, Bennett TM, Hejtmancik JF. Cat-Map: putting cataract on the map. Molecular Vision. 2010; 16: 2007-15. Disponible en: http://cat-map.wustl.edu/.
7. Leysen L, Cassiman C, Vermeer S, Casteels I, Balikova I. Genetics in primary congenital glaucoma: Implications in disease management and counseling. Eur J Med Genet. 2022; 65: 104378. Disponible en: doi.org/10.1016/j.ejmg.2021.104378.
8. Verbakel SK, van Huet RAC, Boon CJF, den Hollander AI, Collin RWJ, Klaver CCW, et al. Nonsyndromic retinitis pigmentosa. Prog Retin Eye Res. 2018; 66: 157-86.
9. Marlhens F, Bareil C, Griffoin J, Zrenner E, Amalric P, Eliaou C, et al. Mutations in RPE65 cause Leber’s congenital amaurosis. Nat Genet. 1997; 17: 139-41. Disponible en: https://doi.org/10.1038/ng1097-139.
10. Chiu W, Lin TY, Chang YC, Lai HIM, Lin S, Ma Ch, et al. An Update on Gene Therapy for Inherited Retinal Dystrophy: Experience in Leber Congenital Amaurosis Clinical Trials. Int J Mol Sci. 2021; 22: 4534. DOI: 10.3390/ijms22094534.
11. Galindo-Bocero J, Macías-Franco S, García-González N, Valles-Antuña C, Hernando Acero I, Rozas-Reyes P. Diagnosis of an X-linked type 2 congenital stationary night blindness using electroretinography and CACNA1F sequencing. Archivos de la Sociedad Española de Oftalmología, 2020; 95: 607-10. Disponible en: https://doi.org/10.1016/j.oftal.2020.06.004.
12. Aboshiha J, Dubis AM, Carroll J, Hardcastle AJ, Michaelides M. The cone dysfunction syndromes. Br J Ophthalmol. 2016; 100: 115-21.
13. Catarino CB, von Livonius B, Priglinger C, Banik R, Matloob S, Tamhankar MA, et al. Real-World Clinical Experience With Idebenone in the Treatment of Leber Hereditary Optic Neuropathy. J Neuroophthalmol. 2020; 40: 558-65. DOI: 10.1097/WNO.0000000000001023.
14.** Holden BA, Fricke TR, Wilson DA, Jong M, Naidoo KS, Sankaridurg P, et al. Global Prevalence of Myopia and High Myopia and Temporal Trends from 2000 through 2050. Ophthalmology. 2016; 123: 1036-42.
15. Mahayana IT, Indrawati SG, Pawiroranu S. The prevalence of uncorrected refractive error in urban, suburban, exurban and rural primary school children in Indonesian population. Int J Ophthalmol. 2017; 10: 1771-6.
16. Yam JC, Jiang Y, Tang SM, Law AKP, Chan JJ, Wong E, et al. Low-Concentration Atropine for Myopia Progression (LAMP) study: a randomized, doubleblinded, placebo-controlled trial of 0.05 %, 0.025 %, and 0.01 % atropine eye drops in myopia control. Ophthalmology. 2019; 126: 113e124.
17. Macías Franco S, Rozas Reyes P. Patología congénita ocular. Pediatr Integral. 2018; XXII: 6-15.
Bibliografía recomendada
– Wang X, Yu C, Tzekov RT, Zhu Y, Li W. The effect of human gene therapy for RPE65-associated Leber’s congenital amaurosis on visual function: a systematic review and meta-analysis. Orphanet J Rare Dis. 2020; 15: 49. DOI: 10.1186/s13023-020-1304-1.
Revisión y metaanálisis sobre los primeros resultados de la terapia génica en amaurosis congénita de Leber.
– Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014; 346: 1258096. DOI: 10.1126/science.1258096.
El trabajo de Charpentier y Doudna explica el mecanismo de las “tijeras moleculares” y edición del ADN. Premio Nobel de Química en el año 2020.
– Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, et al. GeneReviews®. Seattle (WA): University of Washington, Seattle; 1993-2022. What’s New in GeneReviews. Disponible en: https://www.ncbi.nlm.nih.gov/books/NBK132722/.
Recurso con publicaciones en formato revista que incluye buscador y está constantemente actualizándose. Información sobre diagnósticos y asesoramiento genético.
– Shiels A, Bennett TM, Hejtmancik JF. Cat-Map: putting cataract on the map. Molecular Vision. 2010; 16: 2007-15. Disponible en: http://cat-map.wustl.edu/.
Curioso por el formato. Es una base de datos abierta donde se pueden registrar y buscar alteraciones genéticas en pacientes con catarata y asociaciones con enfermedades.
– Holden BA, Fricke TR, Wilson DA, Jong M, Naidoo KS, Sankaridurg P, et al. Global Prevalence of Myopia and High Myopia and Temporal Trends from 2000 through 2050. Ophthalmology. 2016; 123: 1036-42.
Predicción de la prevalencia de la miopía mundial. Publicado en 2016, presenta análisis por subgrupos para detenerse a leerlo.
| Caso clínico |
|
Niña de 10 años que acude con su madre, porque la paciente refiere que no ve la pizarra en clase y le cuesta leer desde hace un año. De inicio pensaron que pudiera padecer dislexia. En la consulta del pediatra, hace 6 meses, la agudeza visual (AV) era de 0,6 en el ojo derecho (OD) y 0,7 en el ojo izquierdo (OI). Además, la paciente refiere que le molesta la luz. Acudieron a una óptica y trae unas gafas de hipermetropía de +0,50 D (dioptrías) que no usa, porque refiere que no mejora con ellas, además nota que cada vez ve peor. En la exploración oftalmológica, la agudeza visual es de 0,5 en el OD y 0,5 en OI que mejora a 0,6 en ambos ojos con corrección subjetiva de +0,50 D. Bajo cicloplejia la corrección es de +1 D, el resto de la exploración es normal, salvo mala colaboración en la realización del fondo de ojo. El segmento anterior y fondo de ojo son normales. Se pauta gafa de +1 D en ambos ojos. A los 6 meses con su gafa de +1 D la AV sigue siendo de 0,5 el segmento anterior y el fondo de ojos normales. Se realiza tomografía de coherencia óptica sin hallazgos.
|

 Ophthalmological emergencies
Ophthalmological emergencies