|
| Temas de FC |
M. Álvarez Molinero*/**, D. Gómez Andrés*
*Neuropediatría. Hospital Universitario Vall d’Hebrón. Barcelona. **Neuropediatría. Hospital Universitario Joan XXIII. Tarragona
| Resumen
Las alteraciones del perímetro craneal son un motivo de consulta y aparecen frecuentemente dudas en su manejo. Tras repasar la metodología de medida y discutir la interpretación de los distintos valores del perímetro cefálico, en el presente trabajo, proponemos los diagnósticos diferenciales en: macrocefalia, microcefalia congénita y microcefalia postnatal, así como algoritmos para relacionar la atención extrahospitalaria generalista con los servicios de Neuropediatría. En estos algoritmos, destacamos la importancia de una interpretación de los valores de perímetro cefálico dentro del contexto clínico y de los hallazgos de la exploración física y del neurodesarrollo, y la incorporación precoz de pruebas genéticas al proceso diagnóstico, con el objetivo de alcanzar un diagnóstico de precisión que permita orientar el cuidado y el pronóstico de manera personalizada en cada paciente. |
| Abstract
Abnormal values of occipitofrontal circumference are a common reason for consultation and uncertainties regarding its clinical management are usually present. This manuscript will review the measurement methods and interpretation of abnormal results, followed by a list of differential diagnosis for macrocephaly, congenital microcephaly and microcephaly, as well as clinical algorithms to support the relationship between primary care and pediatric neurology services. In these algorithms, we have tried to highlight the relevance of an adequate interpretation of abnormal values of occipitofrontal circumference within the clinical context, examination and neurodevelopmental findings. |
Palabras clave: Macrocefalia; Microcefalia; Megalencefalia; Hidrocefalia.
Key words: Macrocephaly; Microcephaly; Megalencephaly; Hydrocephalus.
Pediatr Integral 2020; XXIV (7): 357 – 366
Alteraciones del perímetro cefálico: macrocefalia y microcefalia
La importancia y el dilema de las alteraciones del perímetro craneal(1-3)
El perímetro craneal (cefálico o la circunferencia fronto-occipital) es una medida fácilmente realizable que nos ofrece una información valiosa sobre el desarrollo de los tres componentes del cráneo: parénquima encefálico, líquido cefalorraquídeo y contenido óseo y conectivo.
La medición del perímetro cefálico (PC) debe seguir una técnica. El aparato de medida debe ser una cinta que no se pueda estirar, pero que sea flexible. Es importante buscar la mayor circunferencia posible desde la frente hasta el occipucio. A veces, es útil palpar antes de realizar la medida, para buscar la zona más prominente de la frente (generalmente, 2-4 cm por encima de las cejas) y de la parte occipital. Es importante saber que es frecuente cometer errores de medida, por lo que es conveniente medir varias veces. En caso de alteración, al menos, es importante medir 3 veces.
Como otras tantas cosas en Pediatría, el perímetro cefálico es un fenómeno dinámico. Un recién nacido a término presenta un perímetro craneal de 35 cm (+/-1 cm). Los tres primeros meses de vida el PC puede aumentar 0,5 cm por semana, de los 3 a los 6 meses de vida aumenta 1 cm por mes y, posteriormente, lo hace a un ritmo de 0,5 cm por mes hasta los dos años, completando, de esta manera, el periodo de máximo crecimiento y desarrollo del sistema nervioso. Existen varias curvas de perímetro cefálico cuya principal variación está en los primeros años de vida, por lo que es importante utilizar siempre la misma para describir la evolución y una de las validadas para nuestra población.
El perímetro cefálico es una medida variable dentro de la población general. No existe un punto de corte universal para considerar que un perímetro cefálico es anormal, sino que la definición de micro y macrocefalia depende de otros factores como: la evolución dinámica a lo largo de la edad (no es lo mismo un paciente con +2,5DS de PC que viene desde -1DS o que viene desde +1,5DS), las otras medidas antropométricas, la evolución del neurodesarrollo del niño o los antecedentes médicos y neurológicos del paciente. Por ello, según la fuente bibliográfica, se utilizarán diferentes percentiles o diferentes valores de desviación estándar. En general, podemos establecer una serie de principios a lo hora de interpretar un valor de perímetro cefálico:
• Los pacientes entre -2DS y -3DS son pacientes que, con una alta frecuencia, no van a presentar una alteración neurológica (sobre todo, desde 2DS a 2,5 DS).
• Los pacientes tienen mayor probabilidad de presentar una alteración neurológica cuanto mayor es la desviación de la normalidad. Entre -3DS y +3DS, existe una nula relación entre el perfil cognitivo y la desviación cefálica; pero a partir de esas cifras, el riesgo de dificultades de aprendizaje graves aumenta según aumenta la desviación.
• El error de medida tiene más efecto en términos de percentiles y desviaciones estándar en edades tempranas (por lo tanto, las mediciones de los primeros meses son menos fiables) y es más difícil detectar una desviación franca cuanto más precoz es la medida (p. ej., es más improbable detectar una alteración muy marcada en un neonato que un lactante mayor).
• El perímetro cefálico es una aproximación a lo que desearíamos poder medir que es el volumen cefálico (y, sobre todo, su componente encefálico), que depende de la morfología; es decir, un perímetro redondo se corresponde con mayor volumen que un perímetro elíptico con el mismo valor.
Las alteraciones del crecimiento cerebral pueden ser de causa congénita o adquirida; no obstante, en algunos casos de causa congénita, no se llega al diagnóstico y se pueden clasificar como idiopáticas. Pueden presentarse clínicamente aisladas o constituir parte de síndromes complejos asociados con: otros problemas del neurodesarrollo asociados con: epilepsia, otros problemas del neurodesarrollo (autismo, discapacidad cognitiva u otros), malformaciones cerebrales y anomalías del crecimiento corporal.
Durante la ontogénesis del sistema nervioso central, intervienen una serie de procesos genéticamente programados. Concretamente, en el desarrollo cortical, se suceden los siguientes procesos: proliferación celular, migración, agrupación celular selectiva, citodiferenciación, proliferación axonal (sinaptogénesis) y muerte celular programada (apoptosis). Cualquier disrupción de los procesos biológicos menciados (bien por alteraciones genéticas, infecciones, drogas o disrupciones vasculares, entre otras) puede alterar el desarrollo cortical y, por tanto, el tamaño cerebral.
Es fundamental realizar una correcta anamnesis y una exploración completa. Se debe preguntar sobre los antecedentes pre y perinatales, tales como: infecciones intraútero, exposición a radiaciones, fármacos u otras sustancias tóxicas, edad gestacional, complicaciones durante el parto, sufrimiento fetal, etc. Asimismo, se debe interrogar sobre el desarrollo psicomotor (valorando las tres áreas del desarrollo: motora, social y cognitiva) y si ha habido alguna detención o regresión del desarrollo. Hay que obtener los resultados del cribado neonatal y anotar en la historia clínica, si hay o si no hay signos de hipertensión intracraneal como: irritabilidad, cefalea o vómitos; si tiene dificultades en la visión y audición; antecedentes médico-quirúrgicos; y qué tratamientos toma el paciente. Es importante realizar la anamnesis sobre los antecedentes familiares de interés y medir el PC a los progenitores.
Se debe realizar: exploración física sistematizada, describir el fenotipo y medir la somatometría y constantes vitales. En la valoración neurológica, se debe explorar: morfología craneal, permeabilidad de suturas y fontanelas, y perímetro craneal (desde la zona frontal a la parte más prominente occipital), valorar las funciones cognitivas, explorar los pares craneales, la función motora y sensitiva, los reflejos osteotendinosos, el reflejo cutaneoplantar, la marcha y la manipulación. Siempre que sea posible, hay que realizar un fondo de ojo.
Las alteraciones en el tamaño craneal suelen ser un motivo de preocupación para el pediatra y las familias. En este artículo, pretendemos explicar y proponer un abordaje integral, desde la detección en Atención Primaria hasta las pruebas complementarias avanzadas, que se realizan en la atención especializada.
Macrocefalia(4-7)
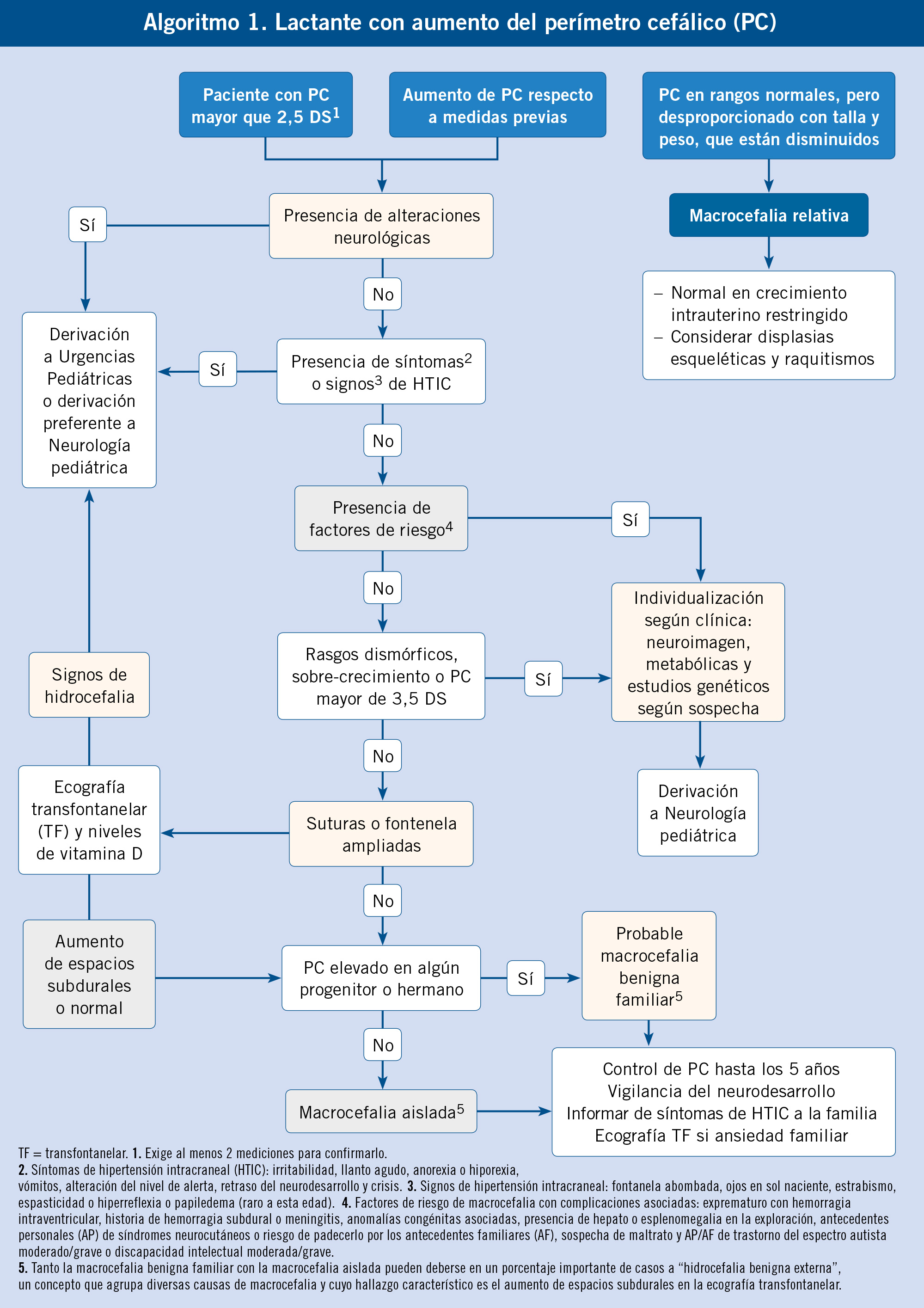
La macrocefalia (perímetro cefálico> 2,5DS) puede deberse a múltiples causas; la exploración física y las manifestaciones de alarma nos ayudan a diagnosticar precozmente causas graves, como la hidrocefalia o las megalencefalias con malformaciones.
Se considera macrocefalia a un perímetro craneal (PC) de más de 2,5 desviaciones estándar (+2,5DS). Los pacientes que presentan un perímetro craneal por encima de 2DS, también se deben tener en consideración en función de la evolución previa. Se define macrocefalia relativa: aquellos pacientes con perímetro craneal en rango normal, pero desproporcionado a su peso y talla, que están disminuidos; es normal en crecimiento intrauterino restringido y se debe descartar raquitismo y algunas displasias esqueléticas, como la osteopetrosis o la osteogénesis imperfecta. En la tabla I, exponemos las principales causas de macrocefalia relativa.

Con megalencefalia, nos referimos al grupo, dentro de las macrocefalias, que se producen por aumento de parénquima cerebral y, por tanto, es un diagnóstico que requiere la realización de algún tipo de prueba de neuroimagen.
El diagnóstico preciso y precoz de la causa de macrocefalia es importante, porque según: etiología, evolución, pronóstico y tratamiento, pueden ser muy distintos. Al final del artículo, proponemos el algoritmo 1 de abordaje para los casos de macrocefalia. El objetivo diagnóstico más importante ante un paciente con macrocefalia, es separar las causas potencialmente graves (Tabla II) de aquellas que tienen un curso benigno.

Los datos claves para distinguir las causas emergentes de las que no son tan urgentes, son las manifestaciones clínicas de hipertensión intracraneal (irritabilidad, cefalea, vómitos, fontanela abombada, ojos en sol naciente o tríada de Cushing, entre otros). Un paciente con manifestaciones compatibles con una hipertensión endocraneal, debe ser remitido a urgencias de Pediatría de forma urgente y deben valorarse con pruebas complementarias para descartar hidrocefalia. En el polo opuesto, están los pacientes con macrocefalia, pero con un desarrollo psicomotor y exploración neurológica normales, y sin antecedentes personales reseñables. En estos casos, un dato clínico relevante es la medición del PC en los progenitores. Si hay antecedentes familiares de macrocefalia, se considerará el diagnóstico de macrocefalia benigna familiar; en caso contrario, hablaremos de una macrocefalia aislada. En ambos casos, precisa vigilancia del PC y control del neurodesarrollo por parte del pediatra de Atención Primaria.
Entre estos dos extremos del diagnóstico diferencial, existe un grupo de pacientes que pueden tener una macrocefalia con datos anómalos en la anamnesis y/o en la exploración. Así, en los casos que el paciente presente: alteraciones neurológicas, factores de riesgo (prematuridad, hemorragia intraventricular, historia de hemorragia subdural, meningitis, anomalías congénitas asociadas, presencia de hepatoesplenomegalia, antecedentes personales o familiares de síndromes neurocutáneos, trastorno del espectro autista moderado/grave o discapacidad intelectual moderada/grave), rasgos dismórficos, síndromes de sobrecrecimiento o perímetro craneal mayor a 3,5 DS, se debe derivar el paciente a la consulta de Neuropediatría. En este grupo de pacientes con factores de riesgo, aunque pueden existir pacientes con hidrocefalia de instauración más lenta, incluimos, sobre todo, megalencefalias. Tradicionalmente, se han dividido en anatómicas (hay un trastorno en la morfogénesis del sistema nervioso central) y en metabólicas (hay un acúmulo anómalo de sustancias). Las megalencefalias suelen tener una base genética y, aunque no siempre se llega al diagnóstico molecular, incluiremos estudios genéticos dentro del protocolo diagnóstico. Se deben solicitar estudios dirigidos en los casos que exista una sospecha diagnóstica evidente, en función de un hallazgo clínico específico. Por ejemplo, si existe macrocefalia y manchas hipocrómicas, se justificaría la secuenciación directa de los genes TSC1/TCS2 en sangre u otro tejido. Si no existe un hallazgo clínico específico, la principal prueba diagnóstica es la RM cerebral con espectroscopia. Es una prueba fundamental, ya que nos da el diagnóstico en el caso de las megalencefalias anatómicas, o nos puede dar la clave para seguir con el diagnóstico, en el caso de las megalencefalias metabólicas. Algunas megalencefalias metabólicas tienen hallazgos radiológicos característicos o patognomónicos que permiten hacer el diagnóstico y confirmarlo directamente con el estudio molecular.
Ante un paciente con megalencefalia, sin antecedentes familiares de megalencefalia, sin alteraciones neurológicas o mínimo retraso del desarrollo con RM con espectroscopia normal o alteraciones mínimas específicas (p. ej., ventriculomegalia), podremos considerar un caso de megalencefalia aislada.
Los pacientes con megalencefalia anatómica suelen nacer con PC alto y la presión intracraneal es normal. Debemos sospechar una megalencefalia anatómica en pacientes con: sobrecrecimiento, dismorfias, trastorno del neurodesarrollo o epilepsia, que cursan de manera relativamente estable (sin regresión). La RM suele mostrar cambios malformativos. Actualmente, la clasificación de las megalencefalias anatómicas se hace basándose en las alteraciones genéticas (Tabla III).

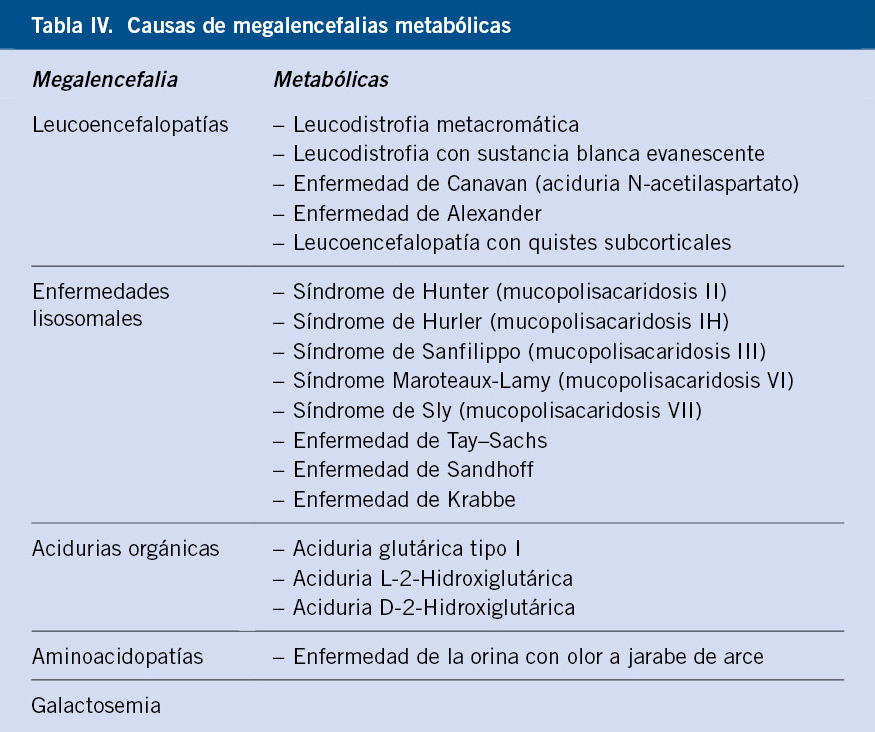
Se conocen diferentes vías moleculares que pueden verse alteradas por hiperactivación en las megalencefalias anatómicas, una de las más conocidas es la vía PI3K-AKT-MTOR. Esta vía molecular es clave en: desarrollo cortical, control de la proliferación celular, migración, citodiferenciación, sinaptogénesis y regulación de la traducción de proteínas; por lo tanto, mutaciones de diferentes genes de esta vía pueden producir una amplia variedad de fenotipos clínicos con: megalencefalia, trastorno del espectro autista, epilepsia y discapacidad intelectual. Algunos de los genes implicados son: PIK3CA, PIK3R2, PTEN, AKT3, TSC1, TSC2, MTOR, CCND2, TBC1D7, RHEB y STRADA. En la tabla III, se resumen las características más importantes de las principales megalencefalias anatómicas. En los casos de sospecha de megalencefalia anatómica o megalencefalia aislada con alteración del desarrollo psicomotor o de la exploración neurológica, en los que no se sospeche una entidad específica, debe solicitar un array CGH y/o WES/WGS (“Whole exome sequencing” / “Whole genome sequencing”). Estas pruebas son importantes para definir el pronóstico de los pacientes y también personalizar su seguimiento. En concreto, ante un paciente con: macrocefalia, regresión importante del desarrollo psicomotor, encefalopatía grave, epilepsia farmacorresistente o alteración grave del neurodesarrollo, y neuroimagen normal, se debe sospechar una megalencefalia metabólica. Se debe realizar un estudio metabólico básico, si este resulta positivo, hay que solicitar estudios metabólicos/genéticos dirigidos. Un factor importante a considerar en las megalencefalias de causa metabólica, es que el PC al nacimiento suele ser normal y la macrocefalia se desarrolla en los primeros meses de vida. En la tabla IV, se exponen las principales causas de megalencefalias metabólicas y, en la tabla V, se resumen aquellas con una neuroimagen característica.

Consideración aparte merecen hallazgos que pueden aparecer en los exámenes y en la neuroimagen de los pacientes con macrocefalia. Por ejemplo, en el caso de que el paciente presente las fontanelas y/o las suturas amplias, se debe solicitar una ecografía transfontanelar (por el riesgo de hidrocefalia), pero también los niveles de vitamina D, por el riesgo de raquitismo. Otros pacientes muestran aumento de espacios subdurales, con un tamaño ventricular normal o mínimamente dilatado (es importante recordar que la atrofia cerebral tendría un volumen ventricular aumentado). En la tabla VI, se exponen las principales entidades que cursan con aumento de espacios subdurales.
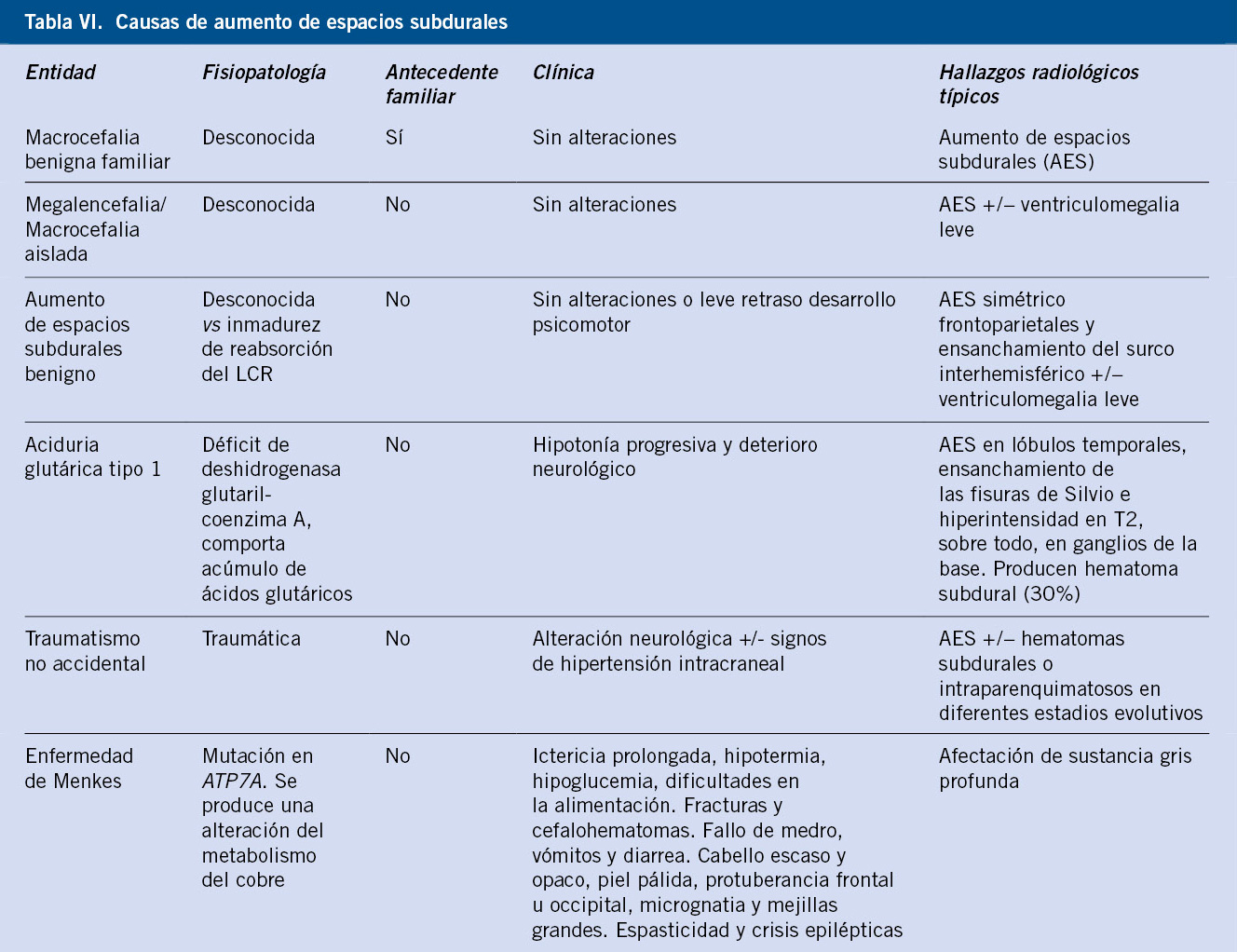
En los pacientes con aumento de espacios, se debe: controlar el perímetro craneal hasta los 5 años de edad, controlar el neurodesarrollo e informar a la familia de signos o síntomas de hipertensión intracraneal, ya que estos pacientes tienen una mayor predisposición a padecer hemorragias subdurales aisladas de forma espontánea o ante un mínimo traumatismo.
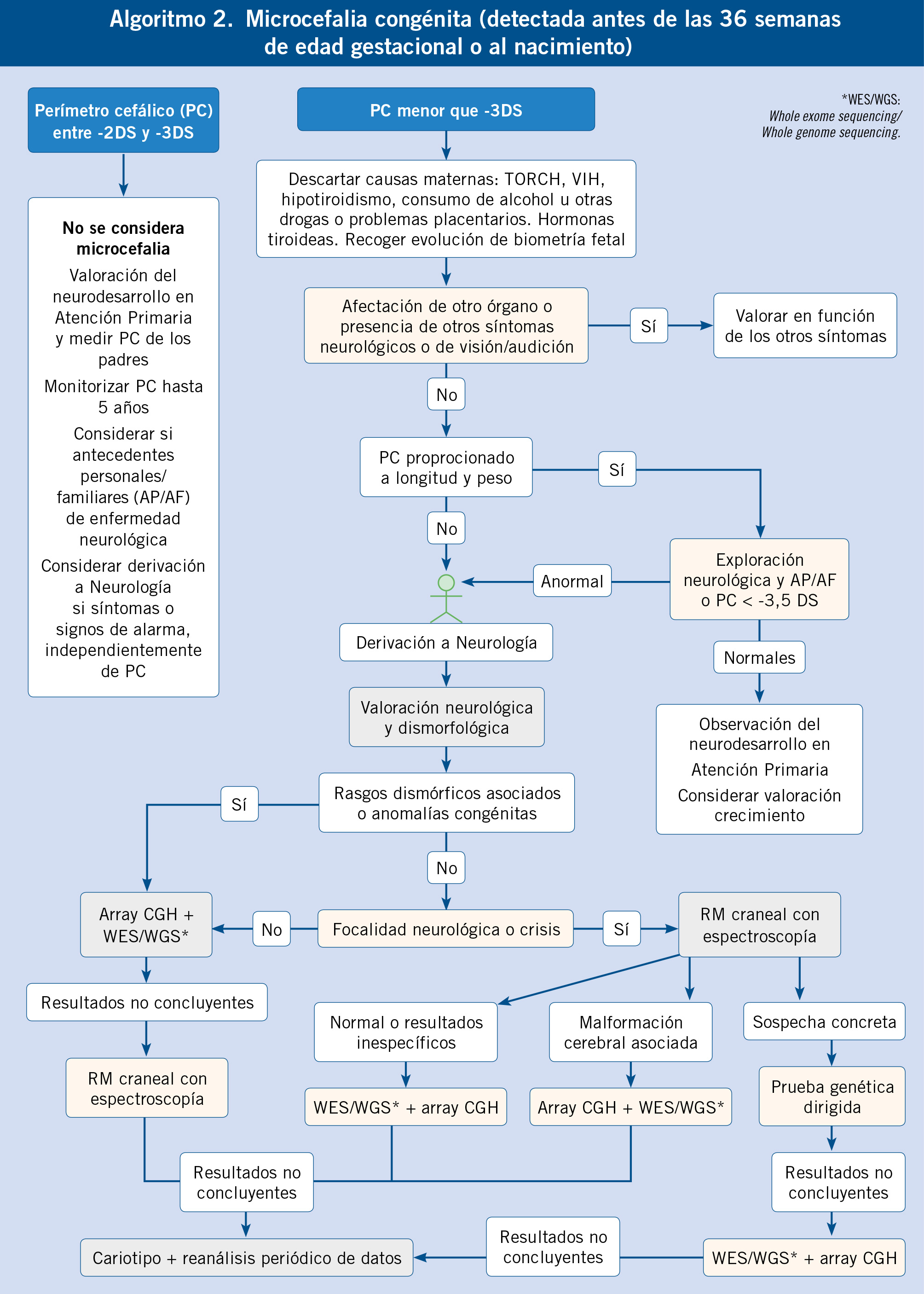
Microcefalia congénita(8-9)
Conceptualmente, dividimos a la microcefalia en dos grandes grupos: microcefalia congénita (pacientes que desde las 36 semanas de edad postconcepcional ya presentan la alteración) y microcefalia postnatal (pacientes en los que la alteración va progresando evolutivamente).
La microcefalia congénita ocurre principalmente por 2 fenómenos:
1. El fallo o reducción de la neurogénesis en las épocas precoces del neurodesarrollo.
2. La alteración de otros elementos del contenido encefálico (sobre todo, sustancia blanca), por un fenómeno disruptivo (infecciones o isquemia intrauterina, exposición a tóxicos…) o un fenómeno neurodegenerativo muy precoz (p. ej., síndrome de Aicardi-Goutières u otras enfermedades genéticas que simulan infecciones congénitas TORCH).
El pronóstico depende de la intensidad de la microcefalia, del momento de la detección (las microcefalias de detección prenatal, antes de las 20 semanas de edad gestacional, tienen un pronóstico mucho peor que las que ocurren posteriormente) y, sobre todo, del diagnóstico concreto de cada paciente y de factores relacionados con este diagnóstico (p. ej., el momento de infección en la infección congénita por CMV).
En el algoritmo 2, al final del artículo, proponemos un abordaje diagnóstico para la microcefalia congénita; pero, desde el punto de vista del diagnóstico diferencial(10), podríamos utilizar los siguientes grupos, sabiendo que existe cierto grado de superposición.
• Microcefalia congénita adquirida: infecciones congénitas (TORCH, VIH y zika), exposición a tóxicos (sobre todo, alcohol y cocaína) o fármacos (en especial, antiepilépticos), y otros factores maternos que condicionen insuficiencia placentaria, anemias graves o malnutrición grave.
• Microcefalia congénita primaria:
– Con anomalías congénitas en otros órganos y/o rasgos dismórficos (microcefalias sindrómicas): orientan hacia síndromes de microdeleción/microduplicación o síndromes monogénicos con afectación multisistémica.
– Con talla baja: es un grupo heterogéneo que agrupa a displasias esqueléticas y a enfermedades con alteraciones en la reparación del ADN.
– Con malformación del desarrollo cortical y/o de la fosa posterior asociada: en este caso, la malformación del desarrollo cortical puede orientar hacia una etiología concreta.
– Con holoprosencefalia asociada: aunque su diagnóstico suele ser sencillo para los neurorradiólogos, las anomalías de la formación de la línea media telencefálica tienen una etiología compleja con implicación de factores ambientales (p. ej.: diabetes materna), cromosomopatías (p. ej.: trisomía 13), enfermedades monogénicas o síndromes de microdeleción/microduplicación.
– Aislada, con patrón giral simplificado y/o aumento leve del tamaño ventricular: son un grupo de enfermedades de herencia autosómica recesiva, cuyo evento patogénico es la reducción de la proliferación de neuroblastos. Tradicionalmente conocidas como: “microcefalia vera”, hoy se prefiere el término de microcefalia primaria hereditaria y, tienen en común, la escasa frecuencia de problemas neurológicos extremadamente graves más allá de: discapacidad intelectual, epilepsia habitualmente no farmacorresistente y leves signos piramidales sin problemas para la marcha autónoma. Sin embargo, existe una importante variabilidad fenotípica asociada a los distintos genotipos; por lo que es recomendable ser reservado desde el punto de vista pronóstico.
La diferencia entre microcefalia congénita adquirida y primaria se realiza basándose en: historia clínica, estudio específico de las causas maternas y, sobre todo, con ayuda de la prueba de imagen. En el caso del diagnóstico diferencial, dentro del grupo de microcefalia congénita primaria, las pruebas genéticas son claves para alcanzar el diagnóstico. En particular, los avances de secuenciación masiva han cambiado el paradigma clásico en el que la neuroimagen todavía jugaba un papel central en todos los pacientes, hacia uno en el que las pruebas genéticas son priorizadas o realizadas al mismo tiempo que la neuroimagen.
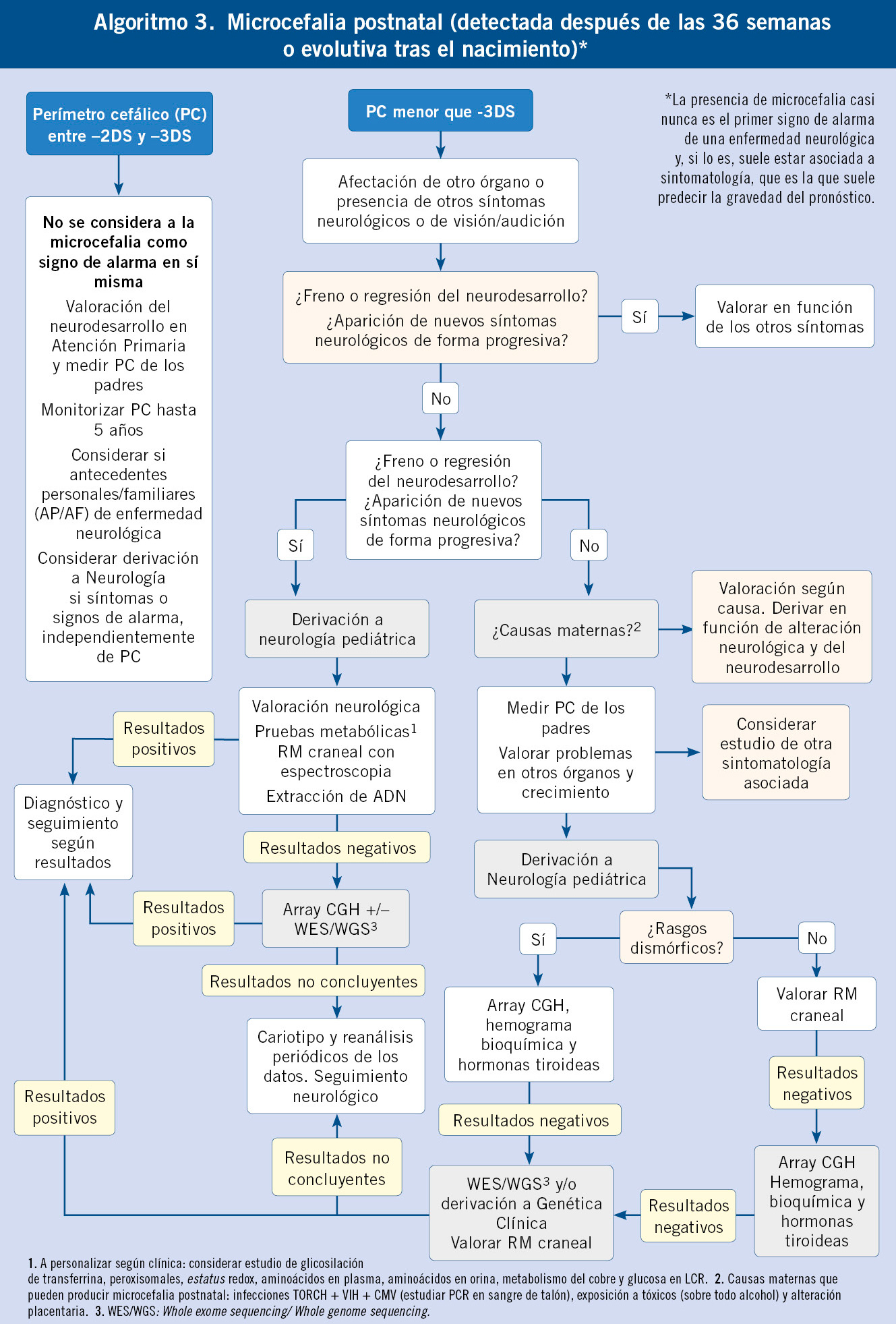
Microcefalia postnatal(9,11)
Bajo este término, agrupamos a todos los pacientes que tienen una disminución del perímetro cefálico que aparece después de las 36 semanas post-concepción o del nacimiento. Dentro del diagnóstico diferencial de esta entidad, es importante separar las causas en las que existe una lesión estática que bloquea el crecimiento normal del sistema nervioso, de aquellas que producen una lesión progresiva. La lesión progresiva generalmente produce más manifestaciones clínicas que la simple microcefalia y, en general, son los síntomas y los signos los que alertan de esta posibilidad. En el algoritmo 3, al final del artículo, incluimos un diagnóstico para el estudio de este grupo de pacientes.
El diagnóstico diferencial de la microcefalia postnatal, en la que se sospecha una lesión estática, incluye: causas adquiridas (encefalopatía hipóxico-isquémica, lesiones adquiridas en relación con la prematuridad, infecciones congénitas…) y causas genéticas. Existe un importante solapamiento entre las etiologías de la microcefalia prenatal y de la microcefalia postnatal, aunque hay enfermedades en las que, aun siendo la lesión relativamente estática, la microcefalia suele ser progresiva o de afectación postnatal, como el déficit de MCT8 (el transportador de hormona tiroidea en el sistema nervioso central).
En el caso de sospecha de patologías progresivas, es importante reseñar alguna de las causas más relevantes como son: enfermedades metabólicas, síndrome de Rett y enfermedades Rett-like caracterizadas por una marcada regresión en torno al año de vida y una afectación grave de las praxias y del lenguaje(12), el síndrome de Angelman y síndromes Angelman-like, el síndrome de Aicardi-Goutières (caracterizado por microcefalia progresiva y encefalopatía subaguda con: brotes de empeoramiento, crisis epiléptica y lesiones cutáneas), las hipoplasias pontocerebelosas o enfermedades que producen también una afectación marcada del crecimiento postnatal, asociada al fenotipo neurológico como: síndrome Warburg MICRO, Cockayne o el síndrome cerebro-oculofacial-esquelético.
Bibliografía
Los asteriscos muestran el interés del artículo a juicio de los autores.
1.*** Piña-Garza JE. Disorders of cranial volume and shape. En: Fenichel’s Clinical Pediatric Neurology. A signs and symptoms approach. Elsevier; 2013. p. 369-84.
2. Woods CG, Parker A. Investigating microcephaly. Arch Dis Child. 2013; 98: 707-13.
3. Forsyth R, Newton R. Head size abnormalities. En: Paediatric Neurology. Oxford specialist handbooks in Paediatrics. Oxford. Oxford University Press; 2012. p. 148-50.
4. Winden KD, Yuskaitis CJ, Poduri A. Megalencephaly and Macrocephaly. Semin Neurol. 2015; 35: 277-87.
5. Pavone P, Praticò AD, Rizzo R, Corsello G, Ruggieri M, Parano E, et al. A clinical review on megalencephaly: A large brain as a possible sign of cerebral impairment. Medicine (Baltimore). 2017; 96: e6814.
6. Orrù E, Calloni SF, Tekes A, Huisman TAGM, Soares BP. The Child with Macrocephaly: Differential Diagnosis and Neuroimaging Findings. AJR Am J Roentgenol. 2018; 210: 848-59.
7. Tan AP, Mankad K, Gonçalves FG, Talenti G, Alexia E. Macrocephaly: Solving the Diagnostic Dilemma. Top Magn Reson Imaging. 2018; 27: 197-217.
8. Pirozzi F, Nelson B, Mirzaa G. From microcephaly to megalencephaly: determinants of brain size. Dialogues Clin Neurosci. 2018; 20: 267-82.
9.*** Woods CG, Parker A. Investigating microcephaly. Arch Dis Child. 2013; 98: 707-13.
10.*** Oegema R, Barakat TS, Wilke M, Stouffs K, Amrom D, Aronica E, et al. International consensus recommendations on the diagnostic work-up for malformations of cortical development. Nat Rev Neurol. 2020. https://doi.org/10.1038/s41582-020-0395-6.
11. Seltzer LE, Paciorkowski AR. Genetic disorders associated with postnatal microcephaly. Am J Med Genet C Semin Med Genet. 2014; 166C: 140-55.
12. Vidal S, Xiol C, Pascual-Alonso A, O’Callaghan M, Pineda M, Armstrong J. Genetic Landscape of Rett Syndrome Spectrum: Improvements and Challenges. Int J Mol Sci. 2019; 20: 3925. https://doi.org/10.3390/ijms20163925.
13. Hinojosa Mena-Bernal J, Pascual B. Trastornos del tamaño y la forma del cráneo. Pediatr Integral. 2015; XIX(9): 591-9.
Bibliografía recomendada
– Piña-Garza JE. Disorders of cranial volume and shape. En Fenichel’s Clinical Pediatric Neurology. A signs and symptoms approach. Elsevier; 2013. p. 369-84.
El capítulo correspondiente en un clásico de la Neurología pediátrica.
– Woods CG, Parker A. Investigating microcephaly. Arch Dis Child. 2013; 98: 707-13.
Magnífica revisión sobre microcefalia. Todo lo esencial está aquí.
– Oegema R, Barakat TS, Wilke M, Stouffs K, Amrom D, Aronica E, et al. International consensus recommendations on the diagnostic work-up for malformations of cortical development. Nat Rev Neurol. 2020. https://doi.org/10.1038/s41582-020-0395-6.
Nuevas guías para el estudio de malformaciones del desarrollo cortical. La propuesta de abordaje genético es interesante.
| Caso clínico |
|
Varón de 6 meses originario de Pakistán, que acude a su primera revisión pediátrica, en la que se detecta un perímetro cefálico de 40 cm (-3,31 DS).
|