 |
| Regreso a las bases |
G. Albi Rodríguez
Profesor asociado del Departamento de Anatomía, Histología y Neurociencia de la Facultad de Medicina de la Universidad Autónoma de Madrid.
Facultativo especialista en Radiodiagnóstico del Hospital Infantil Universitario Niño Jesús de Madrid
Pediatr Integral 2016; XX (7): 485.e1-485.e8
Diagnóstico por la imagen en Oncología Pediátrica (2ª parte)
Introducción
La patología oncológica en Pediatría es muy amplia, más aún, si incluimos las complicaciones derivadas del tratamiento, de modo que exponer los hallazgos de imagen excede los objetivos de este artículo y el previamente publicado. De manera que en este segundo artículo, se van a exponer: tumores de la cresta neural, masas renales y rabdomiosarcoma. Otros tumores, como los óseos se vieron en un artículo previo(1).
La ecografía es la técnica de imagen inicial preferida en Pediatría, sobre todo cuando se sospecha una masa abdominal, ya sea por palpación o por otro síntoma o signo. En ocasiones se podrá demostrar la organodependencia (renal, suprarrenal, etc.) e indicar la prueba de imagen idónea a realizar, generalmente la resonancia magnética (RM), ya que carece de los efectos nocivos de las radiaciones ionizantes de la tomografía computarizada (TC).
Los ultrasonidos también se utilizarán en aquellos casos de masas de partes blandas para determinar su localización y naturaleza (sólida, quística, con estructuras vasculares, etc.), y así, orientar el diagnóstico. De manera que una lesión quística adyacente a una articulación o a una estructura tendinosa será compatible con un ganglión; una masa multiquística podrá corresponder con una malformación vascular de bajo flujo tipo linfática o si se identifican estructuras vasculares podrá ser una malformación venosa. Entre las lesiones sólidas tenemos: pilomatrixomas, tumores dermoides/epidermoides, lipomas, sarcomas de partes blandas, rabdomiosarcomas, etc. Así, la ecografía nos permitirá una primera aproximación para luego realizar una RM, que es la técnica de elección para caracterizar y demostrar la extensión de una masa de partes blandas.
Los tumores derivados de la cresta neural también pueden asentar en el tórax y, a menudo, se diagnostican de forma accidental en una radiografía convencional (Fig. 1). Se utilizará la RM para confirmar su localización en el mediastino posterior y descartar la extensión al conducto raquídeo a través de los agujeros de conjunción.
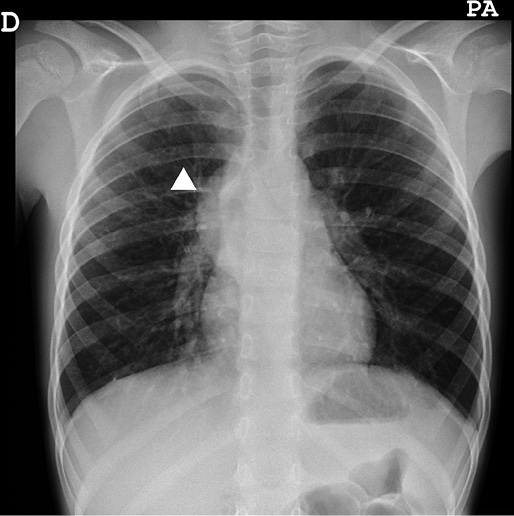
Figura 1. Radiografía de tórax, en la que se visualiza una masa paravertebral derecha que correspondió con un ganglioneuroma (punta de flecha).
Neuroblastoma(2-4)
Es el tumor sólido extracraneal más frecuente en la infancia. Se origina de las células primordiales de la cresta neural, que forman parte del sistema nervioso simpático. Pueden localizarse a lo largo de toda la cadena simpática que discurre paralela a ambos lados de la columna vertebral, desde el cuello hasta la pelvis, aunque también existen células de la cresta neural fuera de esta localización, en el llamado órgano de Zuckerland, situado adyacente a la aorta abdominal distal. Las frecuencias por localización son: adrenales (35%), extraadrenales retroperitoneales (30-35%), mediastino posterior (20%), pelvis (2-3%), cuello (1-5%) y enfermedad metastásica sin primario identificable (1%).
Histológicamente presentan células pequeñas, redondas y azules, similares a otros tumores infantiles, como: sarcoma de Ewing, tumor neuroectodérmico primitivo (PNET, sus siglas en inglés), leucemias, linfomas y rabdomiosarcomas; un signo anatomopatológico clásico es que dichas células forman las llamadas rosetas de Homer Wright.
La presentación clínica depende de la localización anatómica. Las masas abdominales frecuentemente producen dolor, distensión abdominal y son palpables. Pueden comprimir los vasos renales y debutar con hipertensión arterial. Los neuroblastomas torácicos pueden debutar como un síndrome de Horner, con compromiso de la vía aérea, escoliosis o, de forma incidental, en una radiografía de tórax. Otra forma de presentación es detectar múltiples lesiones óseas metastásicas.
El pronóstico es muy variable, debido a sus características biológicas. Incluso se han descrito casos de regresión espontánea. Dependerá de la edad de presentación, el grado histológico, la amplificación del “n-myc”, estadio anatómico. Tienen mayor supervivencia los menores de un año.
El estadiaje ha variado en los últimos años. El “International Neuroblastoma Staging System” (INSS) establecía 5 estadios, incluyendo el 4S (menor de un año; con metástasis hepáticas, piel, óseas; con una supervivencia cercana al 100%), establecidos postquirúrgicamente. Actualmente la estadificación se basa en los hallazgos de imagen previos a la cirugía, según las recomendaciones del “International Neuroblastoma Risk Group” (INRG) propuestas en 2004. Se ha simplificado a 4 estadios (Tabla I), con un papel muy relevante de las pruebas de imagen, ya que se determinan unos factores de riesgo definidos por la imagen, que describen la relación entre el tumor y las estructuras adyacentes (vasculares, de la vía aérea principal y del sistema nervioso central) (Tabla II). Se define que el tumor engloba un vaso cuando lo rodea en el 50% o más, con la excepción de los vasos renales, que cualquier afectación se considera un factor de riesgo definido por la imagen. La ausencia de dichos factores sugiere que la resección será más completa, ya que su existencia conlleva una mayor morbilidad postquirúrgica. Se desconoce si su presencia modifica la supervivencia.


Imagen
Radiología convencional
Aparecen calcificaciones en el 30%.
En los tumores torácicos, en la radiografía de tórax se puede identificar un despegamiento, engrosamiento o irregularidad de las líneas paravertebrales, hallazgo inespecífico, pero muy sugestivo de neoplasia derivada de las células de la cresta neural (Fig. 1). En los niños sanos no suelen ser visibles estas líneas, particularmente la paraespinal derecha, por lo que su engrosamiento es sospechoso de patología en el mediastino posterior. Las masas abdominales que ascienden por el tórax también pueden ser identificadas en una radiografía de tórax en el espacio retrocrural.
Las metástasis óseas se visualizan como lesiones líticas, esclerosas o mixtas.
Ecografía
Suele ser la técnica de diagnóstico inicial, sobre todo en los que se presentan como una masa abdominal. Son lesiones sólidas, heterogéneas por sangrado/necrosis y a veces con calcificaciones, que pueden producir sombra acústica; no siempre dependiente de las glándulas suprarrenales (Fig. 2). Hipervascularizadas en el estudio doppler.
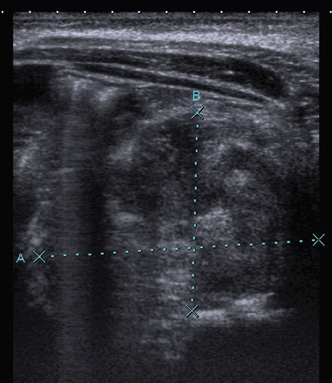

Figura 2. Neuroblastoma extraadrenal, en la ecografía (A), se identifica una masa retroperitoneal de gran tamaño, heterogénea y lobulada. En la RM (B), se demuestra que es independiente de las glándulas suprarrenales, rodea los grandes vasos abdominales y se introduce en el conducto raquídeo por un agujero de conjunción lumbar izquierdo (punta de flecha).
Resonancia magnética
Para la planificación quirúrgica y la estadificación es necesario realizar estudios con técnicas seccionales. Se utilizan la TC o la RM dependiendo, sobre todo, de la disponibilidad. La TC debe hacerse con contraste iodado intravenoso y con equipos multidetector que permiten realizar reconstrucciones multiplanares, con el inconveniente de emplear radiaciones ionizantes. La RM se está convirtiendo en la técnica de elección en muchos centros, y es claramente superior a la TC para detectar la afectación de la médula ósea, la extensión al conducto raquídeo (Fig. 2B) o la invasión de la pared torácica. A parte de la disponibilidad, otra de las limitaciones de esta técnica es la necesidad de anestesia, ya que son exploraciones largas que precisan inmovilidad del niño, generalmente pequeño.
Los neuroblastomas son típicamente hipointensos en T1 y de señal alta en las secuencias potenciadas en T2, y heterogéneas cuando existen calcificaciones, focos de hemorragia o necrosis. El uso de gadolinio intravenoso puede mejorar la visualización de la invasión de los tejidos vecinos. Las secuencias T1 y T2 tienen una excelente resolución de contraste, que permite diferenciar adecuadamente el tumor de las estructuras vasculares vecinas afectadas sin necesidad de contraste intravenoso.
Medicina nuclear
Se utiliza para demostrar enfermedad oculta y la afectación ósea difusa. La gammagrafía con metaiodobencilguanidina es sensible y específica del neuroblastoma cuando se usa en niños. Esta técnica también se emplea en el seguimiento de la enfermedad.
La gammagrafía con Tecnecio-99m se usa para la detección de metástasis óseas.
Se está investigando la utilidad de otros radiotrazadores, como la fluorodesoxiglucosa o el Galio-68 DOTATE (un quelato combinado con un péptido derivado del octreótide).
Diagnóstico diferencial
Un papel fundamental de los métodos de imagen es diferenciar el neuroblastoma adrenal del tumor de Wilms (Tabla III), a veces difícil por el gran tamaño de este tipo de tumores.

Las lesiones en las regiones suprarrenales en un neonato se tienen que distinguir de una hemorragia adrenal, que en la ecografía se aprecia como una masa de pequeño tamaño hipoecogénica, avascular con el doppler, y que se resuelve en los estudios de ultrasonidos de control. Otros tumores adrenales, como el feocromocitoma o el carcinoma adrenocortical, son mucho menos frecuentes en la infancia.
Otras neoplasias derivadas de la cresta neural, como el ganglioneuroblastoma o el ganglioneuroma (Fig. 1), son similares desde el punto de vista de la imagen.
Tumores renales(4,5)
No todos los tumores renales son tumores de Wilms, existe una gran variedad histológica. Su diagnóstico de sospecha puede establecerse por su historia clínica (edad de presentación) y los hallazgos en las técnicas de imagen, de modo que pueda planificarse una estrategia prequirúrgica y establecerse un pronóstico. Aunque no siempre se consigue y hay que recurrir al diagnóstico anatomopatológico. En la tabla IV se muestran las edades de presentación de los tumores renales sólidos malignos más típicos de la infancia:

En la tabla V se resume cómo se visualizan dichas neoplasias en las técnicas de imagen más utilizadas.

Tumor de Wilms
También llamado nefroblastoma. Tercera causa de malignidad en la infancia, tras la leucemia y los tumores del sistema nervioso central. Representa aproximadamente el 87% de todas las masas renales en Pediatría, y es la neoplasia abdominal más frecuente entre 1 y 8 años. Es raro en los neonatos. Puede ser bilateral hasta en el 13% de los niños, y puede estar asociado a anomalías congénitas (criptorquidea, hemihipertrofia, hipospadias y aniridia esporádica). También se puede asociar a determinados síndromes, como el de Beckwith-Wiedemann o el WAGR (Wilms, aniridia, anomalías genitourinarias, retraso mental); a estos pacientes se le realizará seguimiento ecográfico hasta los 7 años, ya que el riesgo de tumor de Wilms a partir de esta edad disminuye significativamente.
Clínicamente puede descubrirse como una masa palpable, a menudo tras un traumatismo. Otros pacientes debutan con síntomas constitucionales y hasta un 25% con hipertensión arterial. La hematuria y el dolor no son muy frecuentes.
Este tumor tiene capacidad metastásica a los pulmones (85%), al hígado y a los ganglios linfáticos locorregionales.
Son tumores grandes, que reemplazan el parénquima renal, que por su tamaño desplazan a las estructuras adyacentes. Se pueden extender a la vena renal y a la vena cava inferior, e incluso alcanzar la aurícula derecha. Pueden producir invasión local.
En las técnicas de imagen son heterogéneos por su contenido hemático, necrosis, grasa o calcificaciones. Cuando se realiza una ecografía y se detecta una masa renal es mandatorio descartar extensión tumoral a la vena cava inferior a través de la vena renal (Fig. 3).
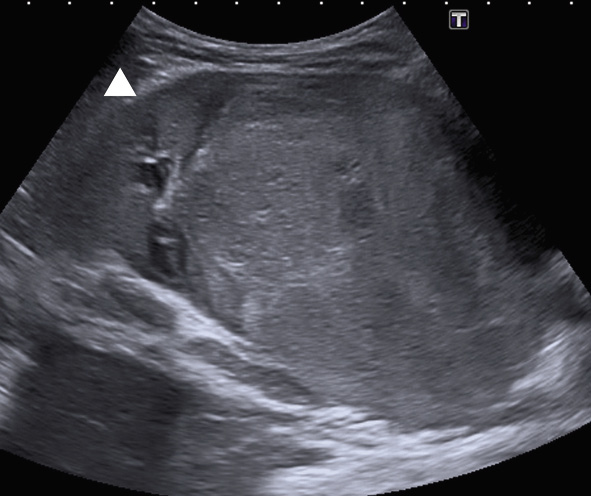
Figura 3. Ecografía de un tumor de Wilms, que se identifica como una masa renal izquierda de gran tamaño, bien definida; se identifica parénquima renal normal (punta de flecha).
Tanto la TC como la RM son usados como complemento a los ultrasonidos en diferentes protocolos oncológicos, dependiendo de su disponibilidad. Aunque la TC no es la técnica ideal por el empleo de radiaciones ionizantes, es necesario que se realice con contraste intravenoso para detectar metástasis hepáticas, afectación ganglionar, invasión vascular, tumores contralaterales sincrónicos y restos nefrogénicos. La RM, además de identificar dichos hallazgos, es más sensible para determinar la permeabilidad de la vena cava, en muchos casos difícil de establecer por la importante distorsión y desplazamiento de distintas estructuras anatómicas debido al gran tamaño de estos tumores en el momento del diagnóstico.
En ocasiones se manifiesta como una gran masa quística, que no se puede diferenciar de un nefroblastoma quístico parcialmente diferenciado, subtipo de un nefroma quístico multilocular.
En el estudio de extensión es obligatoria la TC de tórax para descartar metástasis pulmonares, en cuyo caso no requerirá contraste intravenoso.
El diagnóstico diferencial, desde el punto de vista de la imagen, se establecerá con el neuroblastoma (Tabla III) y con otros tumores renales (nefroma quístico multilocular, sarcoma de células claras, tumor rabdoide renal, nefroblastomatosis, nefroma mesoblástico congénito, angiomiolipoma, carcinoma medular renal y carcinoma de células renales).
Nefroblastomatosis
Consiste en una afectación difusa o multifocal del parénquima renal por restos nefrogénicos. Se pueden detectar hasta en el 1% de autopsias de niños. Aparece desde los neonatos hasta los 7 años. Tiene capacidad de malignización a un tumor de Wilms, más frecuente en pacientes con síndrome de Beckwith-Wiedemann y hemihipertrofia. Se pueden identificar focos de nefroblastomatosis en el 30-40% de dichos tumores. Hay casos en los que hay regresión espontánea de la enfermedad.
Clínicamente pueden ser asintomáticos, debutar como una masa palpable o ser diagnosticados junto con un tumor de Wilms.
En los estudios de imagen son masas sólidas y homogéneas, ovoideas, a veces multifocales y generalmente menores de 3 cm, que suelen situarse en la región cortical del riñón, e incluso bilaterales (Fig. 4).

Figura 4. Múltiples lesiones hipocaptantes en una TC de un niño de 1 año, en relación con nefroblastomatosis bilateral.
El linfoma renal puede ser similar a la nefroblastomatosis, pero no es frecuente en niños pequeños. También hay que diferenciarlo de otras masa renales, aunque suelen ser más heterogéneas.
Nefroma mesoblástico
Es la masa renal sólida más frecuente en los neonatos y en los primeros 3 meses de vida (90% menores de 1 año). Más frecuente en los varones. Se suele manifestar como una masa palpable, la hematuria es poco habitual. Hay casos que se diagnostican intraútero y pueden asociar polihidramnios, hidrops y prematuridad.
La mejor técnica de imagen para su diagnóstico es la ecografía, tanto para el estudio pre- como postnatal. Es imprescindible identificar la glándula suprarrenal normal, que a estas edades se aprecia muy bien, para determinar su origen renal. Se visualiza como una masa intrarrenal, unilateral, sólida, de tamaño variable (desde menos de 1 cm hasta mayores de 15 cm), que afecta típicamente al seno renal y reemplaza a una gran porción del parénquima. Puede contener quistes, focos de hemorragia y de necrosis, aunque estos hallazgos no son habituales. Es frecuente la invasión local del tejido renal adyacente.
Hay que diferenciarlo del tumor de Wilms, del neuroblastoma o ganglioneuroma, de la hemorragia suprarrenal, de la poliquistosis renal autosómica recesiva, de la displasia renal multiquística, del secuestro extrapulmonar y del tumor renal osificante de la infancia.
Tiene un buen pronóstico con la nefrectomía, aunque es necesaria la resección de tejido sano perirrenal dado su capacidad invasiva. La recurrencia local, sobre todo en resecciones incompletas, y las metástasis pulmonares, óseas o cerebrales, son poco frecuentes.
Nefroma quístico multiloculado
Comprende un espectro que va desde una lesión puramente quística (nefroma quístico) hasta una masa con múltiples septos (nefroblastoma quístico parcialmente diferenciado). Los septos son el único componente sólido que presenta este tipo de tumor renal, y que lo diferencia del tumor de Wilms quístico que tiene masa sólida asociada.
Hay dos picos de edad, en niños de 3 meses a 4 años (generalmente, varones con nefroblastoma quístico parcialmente diferenciado) y en adultos (mujeres con nefroma quístico). Se suele manifestar como una masa palpable no dolorosa, hematuria o infección del tracto urinario.
Son masas unilaterales, bien definidas, encapsuladas, con múltiples quistes de varios milímetros hasta 4 cm de diámetro (Fig. 5).
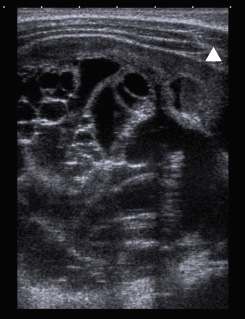
Figura 5. Ecografía de una masa renal formada por múltiples quistes, que correspondió con un nefroma quístico multiloculado, obsérvese el parénquima renal conservado (punta de flecha).
El tamaño total de la masa puede oscilar entre 1 y 30 cm. Cuando se administra contraste en la TC o la RM, existe una captación exclusivamente de las paredes de los quistes. Si estos son todos muy pequeños puede tener una apariencia sólida en estas pruebas de imagen de manera que la ecografía es la mejor herramienta para demostrar su naturaleza quística.
No maligniza, ni se han descrito metástasis, y el pronóstico es excelente tras la cirugía; en los casos excepcionales de recidiva local, la radioterapia o la quimioterapia son tratamientos efectivos.
Sarcoma de células claras
Representa el 4-5% de los tumores renales primarios en Pediatría. Predomina en varones entre 1 y 4 años. Se presenta habitualmente como una masa abdominal. Son lesiones unilaterales indistinguibles por imagen del tumor de Wilms. Tienen mal pronóstico por su agresividad y capacidad para metastatizar al hueso, al hígado, al cerebro, a los ganglios linfáticos y a los pulmones, incluso tiempo después de realizada la nefrectomía.
Tumor rabdoide
Es un tumor exclusivo de niños, aproximadamente el 80% en menores de 2 años y el 60% en menores de uno, la mayoría entre 6 y 12 meses. Es el 2% de todos los tumores renales malignos de la infancia y tiene muy mal pronóstico. Predomina en varones. Clínicamente puede manifestarse como hematuria, pero debido a su agresividad puede debutar como enfermedad metastásica. Puede asociar un tumor rabdoide intracraneal, ya sea sincrónico o metacrónico.
En las pruebas de imagen aparecen como masas grandes, sólidas, heterogéneas, que afectan al hilio renal con márgenes mal definidos, es frecuente la invasión local y vascular.
Angiomiolipoma
Es un tumor renal benigno que no maligniza, raro en la infancia, excepto cuando se asocia a una esclerosis tuberosa. El 20% de los pacientes con este tipo de tumor tienen una esclerosis tuberosa, y el 80% de enfermos con esta entidad tienen angiomiolipomas. También se ha asociado a la neurofibromatosis y al síndrome de von Hippel-Lindau.
Son de tamaño variable, únicos o múltiples, uni o bilaterales.
Pueden ser diagnosticados de forma incidental, ya que la mayoría son asintomáticos, sobre todo, los menores de 4 cm. Cuando son sintomáticos, suelen ser mayores de dicha cifra, y en relación a sangrado intratumoral, con dolor abdominal o en el flanco y hematuria, que puede comprometer la vida del paciente, llamado síndrome de Wünderlich.
La apariencia en imagen varía dependiendo de la cantidad y proporción de sus componentes histológicos. La TC y la RM detectan y caracterizan muy bien aquellos que presentan grasa en su interior, ya que es raro encontrarla en otros tumores renales. La dificultad está en aquellos angiomiolipomas con escaso componente adiposo. Puede ser confundido con un liposarcoma retroperitoneal, aunque es típico del adulto, se ha descrito en adolescentes, simulando un gran angiomiolipoma exofítico.
Rabdomiosarcoma(6-8)
Este tipo de tumor, típico de la infancia, representa el 3-5% de todas las neoplasias malignas de la edad pediátrica. Es el sarcoma de partes blandas más frecuente.
Es una neoplasia de alto grado, se cree que surgen de las células mesenquimales primitivas, y puede ocurrir en múltiples órganos y tejidos, incluyendo aquellos que carecen de músculo estriado.
Aproximadamente el 40% de los casos asientan en la cabeza-cuello, el 15% son genitourinarios no vesicoprostáticos (paratesticulares, vagina, útero, etc.), el 10% son vesicoprostáticos, el 15% en las extremidades, y el 20% en otras localizaciones como el tórax o el abdomen.
La clínica variará dependiendo de la localización.
Tiene capacidad para producir metástasis al pulmón, como lesiones nodulares múltiples, que se diagnostican con la radiografía de tórax o con la TC, siendo esta la técnica de seguimiento.
Rabdomiosarcoma de cabeza y cuello
Es el segundo tumor maligno más frecuente en esta región después del linfoma. La edad media al diagnóstico es entre los 5-6 años, casi el 81% de los pacientes son menores 10 años.
La clínica dependerá de la localización y de la presencia o no de metástasis. Los síntomas pueden ser inespecíficos, como una otitis crónica u obstrucción nasal en los localizados en la región nasofaríngea con cefaleas persistentes. Aquellos que afectan a la base del cráneo pueden debutar como parálisis de pares craneales. Los orbitarios se diagnostican más pronto, ya que tienen síntomas más evidentes, como la proptosis, que también se puede identificar en rabdomiosarcomas de otras localizaciones que invadan la órbita.
La técnica de imagen inicial para valorar una masa de partes blandas, como en otras localizaciones en la infancia, es la ecografía. Aunque en esta región anatómica tiene muchas limitaciones, ya que no puede visualizar adecuadamente los compartimentos profundos del cuello o la base del cráneo, rodeados por hueso o aire. La gran ventaja de los ultrasonidos es que permite una primera aproximación, de forma rápida, a las masas cervicales superficiales, y puede determinar su naturaleza sólida, quística o vascular.
La RM es de elección para demostrar la localización y la extensión. Gracias a su excelente resolución espacial y de contraste, y a su capacidad multiplanar es idónea para la valoración de esta región anatómica tan compleja. Con las antenas adecuadas se puede estudiar la cara, las órbitas, el cuello y la base del cráneo. Además va a permitir determinar la afectación ganglionar locorregional y la diseminación perineural. En los menores de 5-7 años, en la gran mayoría, va a precisar sedación/anestesia, ya que es una exploración que puede durar más de 30-45 minutos. En todos los casos va a ser necesaria la administración de gadolinio, excepto que exista alguna contraindicación.
También es la técnica que se debe utilizar en el seguimiento, para demostrar la respuesta al tratamiento y posibles recurrencias.
La TC, por emplear radiaciones ionizantes, se reserva para determinados pacientes y situaciones.
El diagnóstico diferencial de esta neoplasia se hará dependiendo de la localización anatómica, tanto con lesiones benignas como malignas, por ejemplo con malformaciones vasculares (venosas o linfáticas), tumores benignos de las vainas nerviosas, linfoma, sarcoma de Ewing, osteosarcoma, metástasis, etc.
Rabdomiosarcoma genitourinario
Representan el 25% de todos los rabdomiosarcomas. Se pueden originar de: la vejiga, la vagina, el útero, las paredes de la pelvis, la próstata o de los tejidos paratesticulares. Se subdividen en dos grupos, con diferente pronóstico y tratamiento: los vesicoprostáticos, más desfavorables; y los no vesicoprostáticos (paratesticular, vagina o útero), con mejor pronóstico.
Son masas generalmente grandes, sólidas o quísticas, que tienen un importante efecto de masa sobre los órganos adyacentes, siendo frecuente la obstrucción urinaria. Pueden diseminarse localmente, por vía linfática o hematógena, y producir metástasis pulmonares, hepáticas y óseas.
Existen cuatro tipos histológicos: el de células embrionarias (55%), el alveolar (20%), el indiferenciado (20%) y el botrioide (5%); este último presenta múltiples quistes.
El pico de edad suele ser entre los 2 y los 6 años, siendo el 75% de los casos en menores de 5 años. La excepción es el paratesticular, que es más frecuente en adolescentes.
Clínicamente se manifiestan como una masa abdominopélvica (excepto los paratesticulares intraescrotales) que pueden tener síntomas urinarios (disuria, hematuria, frecuencia miccional, retención urinaria). La presencia de dolor es variable.
La mejor técnica inicial para valorar una masa pélvica o un paciente con síntomas urinarios es la ecografía; luego es necesario una técnica seccional, la más recomendable es la RM, por las mismas razones expuestas anteriormente. Es de elección para descartar una posible extensión al conducto raquídeo. Con todas las técnicas de imagen hay que explorar el sistema excretor urinario y el parénquima hepático.
En el caso del rabdomiosarcoma paratesticular, que se presenta como una masa escrotal, la técnica de elección es la ecografía (Fig. 6) y posteriormente se realizará una TC abdominal para evaluar la extensión ganglionar retroperitoneal.


Figura 6. Se muestran dos imágenes de una ecografía escrotal de un rabdomiosarcoma paratesticular, en la derecha, se aprecia una masa (punta de flecha) de gran tamaño dependiente del testículo izquierdo muy hipervascularizada en el estudio doppler, izquierda.
Rabdomiosarcoma en extremidades
Casi siempre son alveolares, generalmente en niños mayores y adultos jóvenes. Tienen mal pronóstico por su capacidad para metastatizar a los ganglios linfáticos y a localizaciones inusuales.
La técnica inicial es la ecografía, por su disponibilidad y su capacidad para determinar la naturaleza sólida o quística de una masa, y sirve para valorar su vascularización. Hay que recordar que la mayoría de las masas de partes blandas en la infancia son benignas. Se suele identificar como una masa sólida, bien definida, hipoecogénica y discretamente heterogénea (Fig. 7).
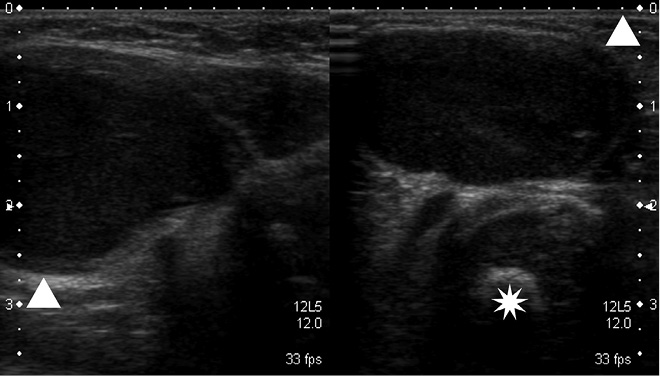
Figura 7. Ecografía de una masa (puntas de flecha) en las partes blandas del hombro (asterisco en la cabeza humeral), sólida, hipoecogénica, bien definida, que correspondió con un rabdomiosarcoma.
La RM es de elección para demostrar la localización (compartimento anatómico), la extensión y su relación con las estructuras vasculonerviosas, información fundamental que necesita el cirujano; también para detectar adenopatías locorregionales.
Bibliografía
1. Albi Rodríguez G. Valoración radiológica de imágenes líticas óseas. Pediatr Integral. 2012; 16: 565-73.
2. Brisse HJ, McCarville MB, Granata C, et al. Guidelines for imaging and staging of neuroblastic tumors: consensus report from the International Neuroblastoma Risk Group Project. Radiology. 2011; 261: 243-57.
3. Nour-Eldin N-EA, Abdelmonem O, Tawfik AM, et al. Pediatric primary and metastatic neuroblastoma: MRI findings Pictorial review. Magn Reson Imaging. 2012; 30: 893-906.
4. Dumba M, Jawad N, McHugh K. Neuroblastoma and nephroblastoma: a radiological review. Cancer Imaging. 2015; 15: 5.
5. Lowe LH, Isuani BH, Heller RM, et al. Pediatric renal masses: Wilms tumor and beyond. Radiographics. 2000; 20: 1585-603.
6. McCarville MB, Spunt SL, Pappo AS. Rhabdomyosarcoma in pediatric patients: the good, the bad, and the unusual. AJR. 2001; 176: 1563-9.
7. Van Rijn RR, Wilde JCH, Bras J, et al. Imaging findings in noncraniofacial childhood rhabdomyosarcoma. Pediatric Radiology. 2008; 38: 617-34.
8. Freling NJM, Merks JHM, Saeed P, et al. Imaging findings in craniofacial childhood rhabdomyosarcoma. Pediatric Radiology. 2010; 40: 1723-38.
