 |
| Temas de FC |
A. Cervera Bravo*, M.T. Álvarez Román**
*Servicio de Pediatría del Hospital de Móstoles, Madrid. **Jefe de Sección de Hemostasia. Hospital Universitario La Paz, Madrid
| Resumen
Se explican las bases fisiológicas de la coagulación, haciendo hincapié tanto en el normal funcionamiento in vivo, como en el modelo clásico de cascada de la coagulación, útil para entender los resultados de las pruebas de cribado de la coagulación. Se habla de los trastornos de coagulación hereditarios más frecuentes: la enfermedad de von Willebrand (EVW), la hemofilia A (déficit de factor VIII) y la hemofilia B (déficit de factor IX). La EVW se debe a la disminución cualitativa o cuantitativa de la actividad del factor de von Willebrand, produce clínica de hemorragia mucocutánea y tiene, generalmente, herencia autosómica dominante. Se muestran las pruebas de laboratorio para diferenciar los distintos subtipos y el tratamiento más adecuado en los mismos. La hemofilia A o B tiene herencia recesiva ligada a X y hay un 30% de mutaciones de novo. La gravedad de la clínica se correlaciona con los niveles del factor. Se expone cómo se realiza el diagnóstico, el diagnóstico diferencial y el tratamiento actual con factor VIII o IX. |
| Abstract
The physiologic basis of coagulation is explained with emphasis on both the normal function in vivo as well as on the classic “cascade” model that better explains the results of coagulation screening tests. The most frequent inherited coagulation disorders are discussed: von Willebrand disease (VWD), hemophilia A (factor VIII deficiency) and hemophilia B (factor IX deficiency). VWD is characterized by a qualitative or quantitative decrease of the von Willebrand factor activity, produces mucocutaneous bleeding and usually has autosomal dominant inheritance. Laboratory work up to differentiate the distinct subtypes and their most adequate treatment are shown. Hemophilia A or B has X-linked recessive inheritance and there is a 30% of de novo mutations. Clinical severity correlates with factor levels. Diagnosis, differential diagnosis and present treatment with factors are presented. |
Palabras clave: Coagulación de la sangre; Enfermedades de von Willebrand; Hemofilia A; Hemofilia B
Key words: Blood coagulation; von Willebrand diseases; Hemophilia A; Hemophilia B
Pediatr Integral 2016; XX (5): 318-330
Fisiopatología y trastornos de la coagulación hereditarios más frecuentes
Fisiología de la hemostasia y coagulación(1,2)
La hemostasia es el mecanismo que se pone en marcha para impedir la hemorragia tras una lesión vascular, en donde participa la pared del vaso sanguíneo, las plaquetas y los factores de coagulación.
Cuando se produce una lesión vascular, se desencadena una respuesta de la pared del vaso dañado con activación plaquetaria y de los factores de la coagulación, que dará lugar a la producción de fibras de fibrina estables unidas firmemente a las plaquetas formando el trombo conjuntamente, que posteriormente, se retraerá por la contracción plaquetaria aproximando los bordes del vaso lesionado para impedir la hemorragia. Las plaquetas segregan factores de crecimiento para las células endoteliales que permitirán la reparación del endotelio dañado. En los bordes de la lesión, donde ya no es necesaria la presencia del trombo, actúan los mecanismos anticoagulantes. Tras la cicatrización y reparación del vaso dañado, se destruye el trombo por fibrinólisis. Si la hemostasia es defectuosa, se producirá una hemorragia. Si es excesiva, porque fallan los mecanismos reguladores, se producirá una trombosis.
Para entender mejor el mecanismo de la hemostasia y coagulación, clásicamente, se la ha dividido en dos fases:
1. Hemostasia primaria: comprende la respuesta vascular, con vasoconstricción inicial por la liberación de adrenalina y la activación plaquetaria. Al exponerse la matriz subendotelial a la circulación, el factor de von Willebrand que va unido en la circulación al factor VIII (FVIII) de la coagulación y el producido por las células contiguas endoteliales se unen a la glicoproteína Ib (GP1b) de las plaquetas activadas y actúa como un pegamento, haciendo que las plaquetas formen como una empalizada de células que tapizan toda la parte dañada (adhesión plaquetaria). Posteriormente, otras plaquetas activadas se unen entre sí, por medio del fibrinógeno a través de las glicoproteína IIb/IIIa (agregación plaquetaria), dando lugar al trombo plaquetario.
2. Hemostasia secundaria (coagulación): es el proceso por el que se activa la cascada de la coagulación, dando lugar a la fibrina estable (Fig. 1).
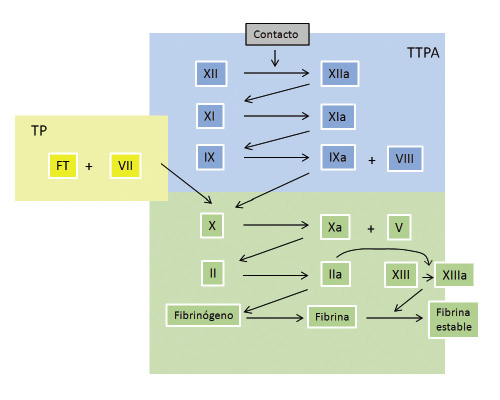
Figura 1. Esquema clásico de la coagulación: la “vía intrínseca” (en azul), evaluada por el tiempo de tromboplastina parcial activado, se inicia al contacto con el vidrio: se activa el factor XII, que junto con sus cofactores kininógeno de alto peso molecular y kalicreína, activan el factor XI y, de ahí, se van activando el resto de los factores hasta la producción de fibrina. La “vía extrínseca” (en amarillo), evaluada por el tiempo de protrombina, se activa al añadir la “tromboplastina tisular” (equivalente del factor tisular FT), que activa el factor VII, este activa el factor X, que a su vez también puede ser activado por el factor IXa de la vía intrínseca, por eso se la llama “vía común” (en verde). Autor de la figura: Áurea Cervera.
Los factores de la coagulación circulan en el plasma como proteínas precursoras inactivas (zimógenos) que, por la acción de una enzima proteolítica que les quita un trozo de la proteína y deja al descubierto su parte activa, se convierten a su vez en enzimas proteolíticas que actúan de igual manera sobre la siguiente proteína de la cascada en una reacción en cadena. Clásicamente, se ha dividido la coagulación en dos vías: la intrínseca y la extrínseca. Esto tiene un sentido didáctico y permite comprender mejor lo que pasa en las pruebas de coagulación que se hacen en el laboratorio y su interpretación (Fig. 1). La llamada vía intrínseca se inicia al contacto de la sangre con las paredes de vidrio del tubo de ensayo que activa la coagulación a partir de los factores de contacto: el factor XII y sus cofactores (kalicreína y kininógeno de alto peso molecular) (Fig. 1). In vivo, la coagulación se inicia por la llamada vía extrínseca (medida por el tiempo de protrombina, TP), cuando al destruirse el endotelio queda expuesto el factor tisular (FT) presente en la membrana de todas las células extravasculares y se pone en contacto con el factor VII circulante (FVII) activándolo. El FVII activado actúa sobre el factor X al igual que lo hace el factor IX (unido a su cofactor, el FVIII) desde la vía intrínseca, por lo que, a partir de ahí, se denomina vía común (Fig. 1). El FX (y su cofactor el FV) activan la protrombina formándose la trombina (IIa). A partir de ahí, se produce una amplificación de la coagulación, porque la trombina además de activar al fibrinógeno para producir fibrina (Fig. 1) y estabilizarla por la activación del factor XIII, activa los factores XI, V, VIII y las plaquetas. Por eso, sangran los hemofílicos, porque no son capaces de amplificar la coagulación tras la formación de la trombina inicial. La coagulación in vivo se lleva a cabo sobre superficies membranosas: las de las células extravasculares expuestas por la lesión endotelial y las de las plaquetas (Fig. 2).

Figura 2.
Esquema de la coagulación in vivo: la coagulación se lleva a cabo sobre membranas biológicas. Se inicia cuando el factor VII se activa al contacto con el factor tisular (FT) de la célula extravascular. Ambos pueden activar al factor X (“vía común”) o al factor IX (“vía intrínseca”), activándose posteriormente los siguientes factores de la cascada hasta la producción de trombina (II). Esta, además de cortar fragmentos del fibrinógeno para que pueda dimerizarse y producir fibrina, amplifica la coagulación activando el factor XI y, desde ahí, al resto de la cascada de la “vía intrínseca” sobre la membrana plaquetaria. Los factores vitamina K dependientes se orientan en la membrana gracias a los puentes de calcio (representados como esferas rojas) que forman con los residuos dicarboxílicos del ácido glutámico.
Anticoagulación:en el borde de la lesión vascular, la célula endotelial contribuye a parar la coagulación: la trombina (IIa) se une a la trombomodulina (TBM) presente en su membrana y activa la proteína C que junto a su cofactor, la proteína S, destruye los factores V y VIII. Además, la proteína membranosa heparán-sulfato, al ponerse en contacto con la antritrombina III circulante, bloquea a los factores II, X, IX, y XI activados. Autor de la figura: Áurea Cervera.
Los trastornos de la hemostasia primaria (enfermedades purpúricas) producen una clínica de hemorragia en mucosas (epistaxis, menorragias, sangrado gingival, etc.) y piel (petequias y equimosis), mientras que los de la hemostasia secundaria (déficit de factores de la coagulación) producen hematomas subcutáneos y profundos (con tumefacción palpable) y sangrado articular (hemartrosis)(3) (Tabla I).

• Anticoagulación: en el borde de la lesión, donde está presente el endotelio sano, ya no es necesario el trombo y se debe limitar la coagulación, por medio de la acción de anticoagulantes. Los principales son la proteína C/proteína S y la antitrombina III. La proteína C se activa por la unión de la trombina a la trombomodulina, proteína presente en la membrana endotelial, y junto a su cofactor, la proteína S, destruye los factores Va y VIIIa. Es decir, que la trombina, en contacto con el endotelio sano, no actúa ya como factor procoagulante sino como anticoagulante (Fig. 2).
• Fibrinólisis: finalmente la restauración de la permeabilidad vascular se establece por la fibrinólisis(4). La fibrina solo puede ser destruida por la plasmina, producida por la activación del plasminógeno unido a la fibrina, que hace que se liberen los productos de degradación de la fibrina, con efecto anticoagulante y los dímeros-D (Fig. 3). Las principales vías estimuladoras e inhibidoras se resumen en la figura 3.
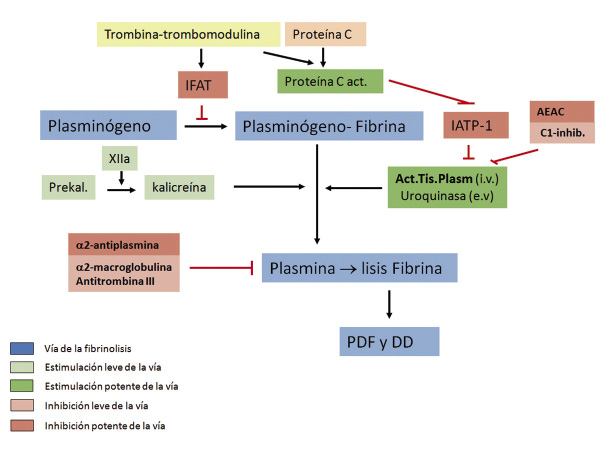
Figura 3. Sistema fibrinolítico. Flechas negras: activación. Barras rojas: inhibición. Prekal: prekalicreína. C1 INH: inhibidor del componente 1 del complemento. IAP-2: Inhibidor del activador tisular del plasminógeno. AEAC: ácido e-amino-caproico. IFAT: inhibidor de la fibrinólisis activable por trombina. PDF: productos de degradación del fibrinógeno. DD: Dímeros D. e.v.: extravascular. i.v.: intravascular. Autor de la figura: Áurea Cervera.
Trastornos hereditarios de la coagulación más frecuentes
Los trastornos de la coagulación más frecuentes son: la enfermedad de von Willebrand, la hemofilia A y la hemofilia B. La mayoría de los trastornos graves se manifiestan en el periodo neonatal o en la primera infancia.
Enfermedad de von Willebrand (EVW)
Es la causa congénita más común de diátesis hemorrágica originada por la deficiencia cualitativa o cuantitativa del factor de von Willebrand (FVW). Tiene herencia autosómica dominante y, menos frecuentemente, recesiva.
La prevalencia en la población por cribado de laboratorio es del 1%(5,6). Existe un trastorno cuantitativo o cualitativo del FVW, proteína multimérica sintetizada en los megacariocitos y células endoteliales, que cumple dos funciones: 1) promover la adhesión y agregación plaquetaria y su unión al colágeno en los lugares de daño endotelial; y 2) actuar como transportador del FVIII, estabilizándolo y prolongando su vida media. El gen de la proteína se encuentra en el brazo corto del cromosoma 12. La proteína que circula en el plasma lo hace con una masa molecular que oscila entre 500-20.000 kDa, según el grado de multimerización de las subunidades. Los multímeros de peso molecular más alto son los que tienen mayor funcionalidad hemostásica. Las manifestaciones hemorrágicas se producen básicamente por la disminución de la actividad del factor de von Willebrand (clínica purpúrica, Tabla I), aunque en algunos casos, pueda sumarse, además, la clínica de los trastornos de la coagulación por la disminución concomitante del FVIII(5).
Clasificación(5-7)
Hay 3 tipos de EVW: el tipo 1 es un déficit cuantitativo parcial del FVW; el tipo 2, déficit cualitativo, consta de cuatro subtipos; el tipo 3 es un déficit grave con niveles muy bajos o indetectables del FVW.
La clasificación de los diferentes tipos y su prevalencia se muestra en la tabla II.

• Tipo 1: es el más frecuente, producido por un déficit cuantitativo parcial del FVW, debido, generalmente, a mutaciones del gen que conllevan una menor síntesis o un mayor aclaramiento del factor, aunque también podría ser consecuencia de la afectación de otros genes que regularan la síntesis del FVW, porque solo es posible detectar alteraciones del gen en 2/3 de los pacientes(8). Es autosómica dominante, pero con gran variabilidad en la penetrancia y expresión clínica.
• Tipo 2: se caracteriza por alteraciones cualitativas del FVW. Se subdivide en 4 subtipos:
- Tipo 2A, tiene un menor número de multímeros de intermedio y alto peso molecular del FVW, que son los más funcionales.
- Tipo 2B, el FVW se une con mayor afinidad a la GPIb, receptor plaquetario del FVW, produciendo una disminución de los multímeros de alto peso molecular y mayor agregación plaquetaria con trombocitopenia en algunos pacientes.
- Tipo 2M, es raro y se caracteriza porque el FVW tiene menor capacidad de unión a la GPIb. Los multímeros están presentes, pero no son funcionales.
- Tipo 2N, es una alteración poco frecuente; la mutación del gen de FVW afecta al lugar de unión del factor VIII, por lo que la función hemostásica del FVW está conservada, pero existe una disminución del factor VIII (5-15% de lo normal) que, al no estar unido al FVW, se cataboliza más rápidamente. Puede confundirse con la hemofilia A.
• Tipo 3: es una forma grave, con afectación de los dos alelos del gen del FVW (homocigotos o dobles heterocigotos), con niveles muy bajos o indetectables de FVW y secundariamente del FVIII.
La herencia, en general, es autosómica dominante, salvo en el tipo 2N, el tipo 3 y algunos casos de otros subtipos, que son autosómicos recesivos (Tabla II).
Clínica(5-8)
La EVW produce clínica de hemorragia mucocutánea exclusiva, salvo los tipos 2N y 3 en los que puede sumarse sangrado en tejidos blandos y profundos.
La EVW de tipo 1 suele pasar bastante desapercibida, ya que en muchos pacientes es una enfermedad leve que produce pocos síntomas de sangrado o estos pasan desapercibidos (menorragias, etc.). Cuando dan clínica, esta es de hemorragia mucocutánea: facilidad de sangrado cutáneo con petequias o equimosis, hemorragias prolongadas de mucosas orofarígea (epistaxis, hemorragias gingivales o tras extracción dentaria, etc.), uterina (menorragias), gastrointestinal (más raro) o de piel (heridas y abrasiones) y tras intervenciones quirúrgicas en dichas localizaciones. Los niños pueden presentar hematomas tras inmunizaciones rutinarias o sangrado gingival por caída de dientes primarios. Algunos pacientes con EVW de tipo 1 con niveles más bajos de FVW, pueden presentar clínica más grave. El tipo 2 cursa con clínica de moderada a grave, salvo el infrecuente tipo 2N, que suele ser leve con sangrado en tejidos blandos, musculares o articulares tras traumatismos, hematuria o hemorragia post-cirugía, parecido a la hemofilia A leve-moderada (ver más adelante), pero con herencia autosómica. El tipo 3 es una forma grave, en la que, a la clínica mucocutánea grave por niveles indetectables de FVW, se suma la de sangrado en los tejidos profundos por disminución severa del FVIII.
La expresión de los síntomas hemorrágicos varía mucho en función de diferentes situaciones que pueden aumentar los niveles del FVW y del FVIII, que son reactantes de fase aguda (estrés, ejercicio, hormonas –estrógenos, hormonas tiroideas–, tratamiento con contraceptivos, etc.), y puede ocultarse la enfermedad(6). Por otro lado, se ha visto que los miembros de una familia con la misma mutación no presentan una clínica hemorrágica similar, porque hay otros factores que pueden contribuir a modular la enfermedad(8,9).
Diagnóstico
El diagnóstico de EVW requiere tres criterios: 1) disminución de la actividad del factor de von Willebrand; 2) clínica hemorrágica; y 3) historia familiar.
El diagnóstico de la EVW no es fácil, por lo que se ha comentado de las diferentes situaciones que pueden aumentar los niveles, o disminuirlos (p. ej.: las personas con grupo sanguíneo O tienen niveles de FVW un 20-30% más bajos). Eso obliga, a veces, a repetir las determinaciones. Por otro lado, los niveles del factor no se correlacionan tan bien con la clínica de sangrado como ocurre con la hemofilia(5,7,8). Además, el diagnóstico de un trastorno hemorrágico debería estar más centrado en la presencia de clínica de sangrado que en los hallazgos incidentales de alteraciones en los resultados de laboratorio. Por eso, en el consenso actual, para el diagnóstico de la EVW, se requiere la presencia de síntomas de sangrado en el paciente y en algún familiar con herencia autosómica dominante o recesiva(5,7).
Es difícil diferenciar los pacientes normales con sangrado de los pacientes con clínica leve con EVW, especialmente en los niños, porque algunas manifestaciones clínicas hemorrágicas, como las epistaxis o las equimosis (moratones), son muy frecuentes en niños sanos(10,11). Además, en los enfermos pediátricos, la historia de sangrado puede ser difícil de obtener, porque todavía no les ha dado tiempo a enfrentarse a situaciones de riesgo (cirugías, extracciones dentales, etc.)(5). Por eso, el requerimiento diagnóstico de que exista clínica hemorrágica en los niños es más controvertido(10)y un correcto diagnóstico en ellos requiere repetidas determinaciones de los niveles de FVW junto con una adecuada historia familiar(5).
En los últimos años, se ha desarrollado como cribado de enfermedad hemorrágica, una escala de sangrado con una lista de preguntas estandarizadas unidas a un puntaje(12) (Tabla III), que intentan diferenciar los pacientes sanos de los enfermos y que se correlaciona con la gravedad de la EVW.

Las preguntas más informativas están relacionadas con el sangrado prolongado tras la cirugía o extracciones dentales, la presencia de hematomas musculares o hemartros, y la historia familiar de una enfermedad hemorrágica establecida(7). En los adultos, esta escala incluso predice mejor el riesgo de posible sangrado tras intervenciones quirúrgicas o extracciones dentales que los niveles del factor(9). La escala de sangrado ha sido adaptada a los niños(13), pero tiene menos sensibilidad para detectar los enfermos, aunque el factor predictivo negativo es muy alto y podría ser útil para su empleo en Atención Primaria para valorar qué pacientes deben ser referidos a estudio(11).
Las pruebas analíticas deben iniciarse con un cribado de la coagulación (Algoritmo 1). Puede haber una disminución del FVIII coagulante (FVIIIc) paralela a la disminución cuantitativa del FVW que prolongue levemente el tiempo de tromboplastina parcial activado (TTPA). El PFA-100® o tiempo de obturación puede ser un buen método de cribado(5) (mide el tiempo que tarda en cerrarse la abertura de un cartucho recubierto de colágeno con ADP o epinefrina al pasar por ella sangre citratada: las plaquetas tienden a adherirse y agregarse cerrando el orificio). En la EVW (al igual que en las alteraciones plaquetarias) está prolongado; sin embargo, en algunos pacientes puede ser normal, por lo que algunos autores no lo recomiendan para descartar la EVW o problemas de la función plaquetaria(3). El tiempo de hemorragia es menos sensible y es más difícil de estandarizar(3,6,7), por lo que ya no se utiliza. Por tanto, si la clínica y los antecedentes familiares son sugerentes, se debería estudiar directamente el FVW(7).
El estudio del FVW incluye los niveles cuantitativos de la proteína (FVW:Ag), su función o actividad por la prueba del cofactor de ristocetina (FVW:RCo) y los niveles del FVIIIc. Niveles bajos de FVW:Ag entre un 35-50 UI/dL pueden aparecer también en personas normales y solo se asocian a anomalías del gen en un 51% de los casos(7), por lo que el umbral del nivel a considerar como patológico, también es controvertido(5,6,10). Se considera como patológico <30%, y niveles bajos (que pueden corresponder tanto a individuos sanos como enfermos) entre 30-50%. El diagnóstico de los diferentes subtipos 2 requiere estudios especializados (algoritmo 1).
Al diagnóstico, se debe incluir en todos los pacientes con el tipo 1, la respuesta a la desmopresina (1-desamino-8-D-arginina vasopresina: DDAVP), ya que será el tratamiento a emplear en la mayoría de los pacientes, aunque no todos responden.
Diagnóstico diferencial
Se debe diferenciar de los problemas plaquetarios y de las formas graves de la hemofilia.
La EVW puede manifestarse clínicamente de forma similar a los problemas plaquetarios (disminución cuantitativa o problemas de la función plaquetaria). El subtipo 2B puede confundirse clínicamente con la enfermedad de Bernard-Soulier, porque ambas presentan trombopenia leve, aunque en esta última enfermedad, el problema está en la ausencia de GP1b, las plaquetas son gigantes y los niveles de FVW son normales.
La prolongación aislada del TTPA en las pruebas de cribado de coagulación debe hacer sospechar una EVW si existe clínica de sangrado mucocutáneo o historia familiar. Cuando la clínica es de hematomas profundos o hemartros, debe sospecharse una hemofilia o, mucho menos frecuente, la EVW 2N (p. ej.: niñas). Si no existe clínica de sangrado, la mayoría de los pacientes suelen tener diagnósticos sin ninguna relevancia clínica (como anticoagulantes lúpicos transitorios, frecuentes tras infecciones virales, el déficit de factores de contacto, etc.). En este grupo, el diagnóstico de EVW es mucho más raro, aunque en un estudio de nuestro medio representaban el 15%(10).
Tratamiento
El tratamiento fundamental en la EVW tipo 1 y el tipo 2 leve es la desmopresina; en los tipos 2 moderados-graves, o en el tipo 3, los concentrados de FVW. Los antifibrinolíticos son útiles como tratamiento coadyuvante.
En todos los pacientes con EVW, excepto en el tipo 2B o el tipo 3, se debe ensayar al diagnóstico la respuesta a la desmopresina (1-desamino-8-D-arginina vasopresina: DDAVP), que libera tanto el FVIII como el FVW de su lugar de almacenamiento en el endotelio. La respuesta es variable de unos pacientes a otros, pero es consistente en un mismo paciente y permite planificar el tratamiento más adecuado en las diferentes situaciones clínicas (hemorragias o pre-cirugía). La mayoría de los pacientes con tipo 1 responden y un porcentaje de los de tipo 2, generalmente los menos graves, aunque se debe ensayar en todos(14,15) (Algoritmo 2). En la mayoría de las formas graves del tipo 2, el defecto cualitativo no puede ser compensado por la liberación de más moléculas de FVW defectuoso. En el tipo 2B, la liberación de FVW anormal puede aumentar la aglutinación plaquetaria y acentuar la trombopenia, por lo que generalmente no se recomienda(14,15), aunque en algunos pacientes puede funcionar(15); en el tipo 3 no habrá respuesta. La dosis y administración se muestra en la tabla IV.

Produce un incremento de 3 a 5 veces de los niveles basales hacia los 30-60 minutos y dura de 6-12 horas. Tiene efecto de taquifilaxia (disminución de la acción con las dosis repetidas), no se recomienda más que una vez al día y, en niños, la hiponatremia puede producir convulsiones, por lo que no se recomienda en niños de menos de dos años si no hay una unidad de vigilancia intensiva. Se debe monitorizar la actividad del FVW:RCo y del FVIII en casos de cirugía mayor, pero no es necesaria para episodios de sangrados leves. Por el fenómeno de taquifilaxia, si se precisa mantener niveles adecuados de FVW más de 3 días, hay que emplear concentrados de FVW. Puede aparecer: enrojecimiento, cefalea, hiper o hipotensión, molestias gastrointestinales, hiponatremia y hormigueo como efectos secundarios(7) (Tabla IV). La administración intranasal es más práctica, porque permite hacerlo de forma ambulatoria antes de procedimientos menores, como extracciones dentales, etc. Hay que tener en cuenta, que la presentación intranasal recomendada en la EVW tiene una concentración tres veces mayor que la que se utiliza en el tratamiento de la enuresis o la diabetes insípida. No se recomienda dar anti-inflamatorios no esteroideos en la EVW, porque alteran la función plaquetaria. Su empleo junto con el DDAVP puede acentuar la hiponatremia. La restricción de líquidos disminuye el riesgo de hiponatremia(15).
En los casos en los que no puede administrarse el DDAVP, hay que emplear concentrados plasmáticos de FVIII/FVW inactivados para virus, que contengan FVW. Existen unas recomendaciones para las dosis a utilizar dependiendo del tipo de hemorragia o como profilaxis en cirugía, para mantener niveles adecuados de FvW:RCo y FVIII(14,15) (Tabla IV).
El tratamiento con antifibrinolíticos (Tabla IV) puede ser eficaz como tratamiento aislado o adyuvante en todos los pacientes con sangrado en mucosas con gran actividad fibrinolítica (epistaxis, extracciones dentarias, menorragias, etc.) al estabilizar el coágulo(7,14). Está contraindicado en la hematuria, porque podría producir obstrucción de la vía urinaria. El tratamiento con estrógenos puede emplearse en las menorragias que no responden a antifibrinolíticos o DDAVP intranasal(14). La aplicación local de agentes tópicos (selladores de fibrina, colágeno, trombina tópica) puede restablecer la hemostasia en vasos pequeños, especialmente en la cavidad bucal o nasal(7,15).
Hemofilia A y B
Se originan por el déficit o ausencia de la función del factor VIII (hemofilia A) o IX (hemofilia B). Tienen herencia recesiva ligada al X.
La hemofilia A es aproximadamente 5-6 veces más frecuente que la B, afecta a 1/5.000 recién nacidos varones, frente a la B que lo hace a 1/30.000-50.000. Además, es grave con mayor frecuencia: en dos tercios de los pacientes con hemofilia A y en un 50% de los que tienen hemofilia B. Se producen por mutaciones de los genes del FVIII o del FIX en el cromosoma X. Un tercio de las mutaciones aparecen de novo, dando lugar a una mujer portadora (más frecuentemente) o un varón enfermo, en los que no habrá antecedentes familiares. La clínica es idéntica, ya que ambos factores actúan conjuntamente en la vía intrínseca de la coagulación(16).
Clínica
Los niveles de actividad del factor determinan la gravedad clínica del cuadro: hemofilia grave <1% de actividad; moderada: actividad del 1-5%; leve ≥ 5% de actividad.
La actividad del factor se define en unidades: una unidad internacional es la cantidad presente de factor en 1 ml de un pool de plasma normal (1%). El nivel de actividad del factor define la gravedad de las manifestaciones clínicas y la edad de aparición de la enfermedad. Aunque los pacientes hemofílicos pueden sangrar en cualquier área, son las hemorragias en las articulaciones y los hematomas musculares los más específicos y los que producen, sin tratamiento adecuado, una mayor morbilidad. Los casos graves presentan sangrado profundo de forma espontánea o con mínimos traumatismos y, generalmente, los síntomas aparecen antes del primer año de vida, cuando inician la deambulación, aunque en un 2% puede aparecer ya en el recién nacido, generalmente como hemorragia cerebral. Los moderados tienen menos hemorragias espontáneas, pero pueden sangrar de forma importante tras traumatismos o cirugía y dan la cara más tarde. Los leves sangran solo con traumatismos o tras cirugía(16) (Tabla V).

Las formas habituales de presentación en los primeros años son: equimosis, hematomas grandes al mínimo traumatismo (p. ej., vacunaciones intramusculares, hematomas frontales) y sangrado bucal (p. ej., rotura de frenillo). Un 30% sangran en la circuncisión. La hemartrosis puede presentarse en cualquier articulación, pero el tobillo, la rodilla y el codo suelen ser las localizaciones más frecuentes. Se manifiesta como: dolor, tumefacción y aumento de la temperatura de la articulación, con impotencia funcional, con o sin traumatismo previo. Las hemartrosis repetidas producen una hipertrofia sinovial, que facilita nuevos sangrados, depósitos de hemosiderina, sinovitis crónica y, en pocos años, se desarrolla una artropatía grave irreversible(16). Los niños mayores son capaces de detectar el sangrado precozmente por una sensación de hormigueo y calor en la articulación(1). Los hematomas musculares, especialmente en el psoas-ilíaco (que puede dar solo un dolor inguinal vago, pero puede llevar a un shock hipovolémico por la pérdida de grandes volúmenes de sangre) o en el brazo, con compromiso vascular, y las hemorragias del SNC son otras de las manifestaciones clínicas graves que precisan tratamiento sustitutivo inmediato(1,16). Los pacientes con déficit moderado o leve suelen diagnosticarse más tarde y, en ocasiones, se detectan en análisis preoperatorios. Algunas mujeres portadoras pueden presentar síntomas de sangrado por el fenómeno de Lyonización (como una hemofilia leve)(1,16).
Diagnóstico. Diagnóstico diferencial
El diagnóstico se establece por la disminución de la actividad del factor.
El déficit de FVIII o FIX produce una prolongación del TTPA con tiempo de protrombina normal. La confirmación se da cuando el nivel de actividad del factor es inferior al normal en dos determinaciones analíticas (<50% o 50 UI/dL), tanto por déficit de la proteína, como por alteración de su función. En todas las portadoras, se deben medir los niveles del factor antes de procedimientos quirúrgicos, para valorar el tratamiento(1). Hay que tener en cuenta que, aunque los niveles de FVIII son ya como los del adulto en el neonato a término, los de FIX tardan más en alcanzarlos, por lo que no debe diagnosticarse una hemofilia B leve antes del año de edad por estudio de factores(1).
El diagnóstico diferencial principal se establece con la EVW, porque, como se ha comentado, los niveles de FVIII en la EVW pueden ser bajos. Por eso, es necesario también determinar los niveles y función del FVW. En los lactantes con sangrado grave, el diagnóstico diferencial se establece con trombopenias graves o alteraciones graves de la función plaquetaria, como el síndrome de Bernard-Soulier o la tromboastenia de Glanzmann, aunque el hemograma y el cribado de coagulación diferenciarán estas entidades(1).
Tratamiento
El tratamiento principal es el empleo de concentrado plasmático inactivado o recombinante de forma precoz en las hemorragias y de forma profiláctica (casos graves). En la hemofilia A moderada o leve, se puede emplear el DDAVP.
El tratamiento se basa en el empleo de FVIII o FIX recombinante, que evita la transmisión de posibles agentes infecciosos, como ocurrió en el pasado, al no ser un producto biológico de origen humano y tener una muy escasa o nula contaminación proteica y celular humana o animal. Los derivados de origen plasmático también tienen un alto grado de seguridad por el proceso de inactivación viral(17,18), aunque podrían transmitir parvovirus, hepatitis A y priones(16). Una unidad del concentrado/kg sube la actividad al 2% de FVIII y alrededor del 1% de FIX. Los niveles hemostáticos para el FVIII son de 30-40% y de 25-30% para el FIX(1). La vida media es de 8-12 horas para el FVIII y de unas 17 horas para el FIX. El nivel de hemostasia al que se debe llegar depende del tipo y gravedad de la hemorragia (Tabla IV). Se debe emplear de forma precoz en las hemorragias para evitar mayor morbilidad.
El mayor avance en los últimos años ha sido el empleo profiláctico de factor en los casos graves, con infusiones 3-4 veces/semana de FVIII o dos/semana de FIX a partir de 1-3 años, porque eso evita el desarrollo de la artropatía hemofílica, convirtiendo a los hemofílicos graves en moderados, lo que se ha demostrado que es coste-eficaz(17). Para ello, se necesita la implantación de una vía central de acceso venoso para poder recibir el tratamiento domiciliario profiláctico o precoz en las hemorragias(17), aunque a su vez, la presencia de vías centrales tiene complicaciones(18). El mayor problema del tratamiento profiláctico es el desarrollo de inhibidores (anticuerpos frente al FVIII o FIX) que aparecen en un 20-30% de los pacientes graves con hemofilia A y en un 5% de los que tienen hemofilia B, generalmente por una predisposición genética(17). La profilaxis secundaria en niños mayores y adolescentes con artropatía ya instaurada, también podría ser eficaz(17,18). Se están desarrollando moléculas modificadas de factores VIII y IX con vidas medias más prolongadas que permitirán reducir el número de inyecciones(17,18). Además de la profilaxis, se debe infundir el factor lo antes posible con la aparición de hemartrosis o hematomas profundos para evitar daño irreparable.
En los hemofílicos A moderados a leves, se puede emplear el DDAVP asociado o no a antifibrinolíticos (Tabla IV) y otras medidas coadyuvantes ya comentadas en la EVW. El DDAVP en la hemofilia B es ineficaz.
Las vacunas rutinarias deben administrarse por vía subcutánea profunda o por vía convencional con la aguja más pequeña posible y aplicando posteriormente presión y frío local. Se recomienda vacunar además de la hepatitis A(17,18). Los AINES (aspirina, ibuprofeno) están contraindicados. Se debe emplear para el dolor paracetamol o el metamizol y en mayores de 12 años se podría emplear además la codeína.
Es importante potenciar la actividad física y vigilar el estado nutricional, para no contribuir al sobrepeso y a la limitación de movimientos a los que son especialmente vulnerables(18).
Función del pediatra de Atención Primaria
• Sospecha diagnóstica de un trastorno hereditario de la coagulación: realizar historia clínica y una exploración física cuidadosas, diferenciando sangrado en niño sano (epistaxis o equimosis con estudios de hemostasia normales, sin historia familiar) de enfermo (sangrado significativo, historia familiar, pruebas alteradas de hemostasia). Si la clínica de sangrado o la anamnesis familiar es significativa, aunque el cribado de coagulación sea normal habría que derivar al paciente para cuantificar el factor.
• Administración de vacunas habituales por vía subcutánea con presión y frío local en enfermos con clínica moderada-grave. Administrar además la vacuna de VHA en todos los que precisen factor.
• Evitar la administración de antiinflamatorios no esteroideos en pacientes moderados-graves.
• Conocer las bases de sus tratamientos, en especial, el empleo de DDAVP intranasal en los que esté indicado y los tratamientos adyuvantes (esteroides, antifibrinolíticos…) para su empleo en procedimientos dentales o de cirugía menor.
• Recomendaciones sobre alimentación, higiene dental, ejercicio (en colaboración con hematólogo).
Bibliografía
Los asteriscos reflejan el interés del artículo a juicio del autor.
1.** Scott JP, Raffini LJ, Montgomery RR, Flood VH. Hemorrhagic and thrombotic diseases. In: Kliegman RM, Stanton BF, St Gemme JW, Schor NF, Behrman RE, eds. Nelson Textbook of Pediatrics, 20 ed. Elsevier; 2016. p. 2379-408.
2. Carrillo J. Hemostasia y coagulación. II Curso de Diagnóstico Integral en Hematología. Módulo 9. Madrid. Hospital Universitario Ramón y Cajal, 29-30 de enero de 2016.
3.* Yee DL, Mahoney DH, Armsby C. Approach to the child with bleeding symptoms. UpToDate. Versión Enero 2016. Actualizado el 29/07/2015. Consultado el 22/02/2016. Disponible en: www.uptodate.com.
4. Chapin JC, Hajjar KA. Fibrinolysis and the control of blood coagulation. Blood Rev. 2015; 29: 17-24.
5.** Federici AB. Clinical and laboratory diagnosis of VWD. Hematology Am Soc Hematol Educ Program. 2014; 1: 524-30.
6.** Rick ME, Leung LLK, Timauer JS. Clinical presentation and diagnosis of von Willebrand disease. UpToDate. Versión Enero 2016. Actualizado el 16/08/2013. Consultado el 22/02/2016. Disponible en: www.uptodate.com.
7.** Nichols WL, Hultin MB, James AH, et al. von Willebrand disease (VWD): evidence-based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) Expert Panel report (USA). Haemophilia. 2008; 14: 171-232.
8.* Flood VH. New insights into genotype and phenotype of VWD. Hematology Am Soc Hematol Educ Program. 2014; 1: 531-5.
9. Federici AB, Bucciarelli P, Castaman G, et al. The bleeding score predicts clinical outcomes and replacement therapy in adults with von Willebrand disease. Blood. 2014; 123: 4037-44.
10. Romero L, Conde N, García Aldana D, Ruano A, Fernández-Tejeiro A. Revisión de los pacientes estudiados por coagulopatía en una unidad de Oncohematología. An Pediatr (Barc). 2016; 84: 85-91.
11. Mittal N, Pedersen R, James P, Shott S, Valentino LA. Utility of a Pediatric Bleeding Questionnaire as a screening tool for von Willebrand disease in apparently healthy children. Haemophilia. 2015; 21: 806-11.
12. Tosetto A, Rodeghiero F, Castaman G, et al. A quantitative analysis of bleeding symptoms in type 1 von Willebrand disease: results from a multicenter European study (MCMDM-1VWD). J Thromb Haemost. 2006; 4: 766-73.
13. Bowman M, Riddel J, Ransd ML, et al. Evaluation of the diagnostic utility for von Willebrand disease of a pediatric bleeding questionnaire. J Thromb Haemost. 2009; 7: 1418-21.
14.** Rodeghiero F, Castaman G, Tosetto A. How I treat von Willebrand disease. Blood. 2009; 114: 1158-65.
15.** Neff AT, Sidonio RF. Management of VWD. Hematology Am Soc Hematol Educ Program. 2014; 1: 536-40.
16.** Hoots KW, Shapiro AD, Leung LLK, Mahoney DH, Tirnauer JS. Clinical manifestations an diagnosis of hemophilia. UpToDate. Versión Enero 2016. Actualizado el 11/11/2014. Consultado el 26/02/2016. Disponible en: www.uptodate.com.
17. Coppola A, Morfini M, Cimino E, Tufano A, Cerbone AM, Di Minno G. Current and evolving features in the clinical management of hemophilia. Blood Rev. 2014; 12(suppl 3): s554-62.
18.* Hoots KW, Shapiro AD, Mahoney DH, Tirnauer JS. Treatment of hemophilia. UpToDate. Versión Enero 2016. Actualizado el 3/08/2015. Consultado el 26/02/2016. Disponible en: www.uptodate.com.
Bibliografía recomendada
- Scott JP, Raffini LJ, Montgomery RR, Flood VH. Hemorrhagic and thrombotic diseases. In: Kliegman RM, Stanton BF, St Gemme JW, Schor NF, Behrman RE, eds. Nelson Textbook of Pediatrics, 20 ed. Elsevier; 2016. p. 2379-408.
Revisión bastante completa y actualizada, sin ser exhaustiva, ideal para un pediatra general, de la fisiología de la coagulación, estudios de laboratorio y principales enfermedades hemorrágicas y trombóticas en la infancia, con tablas de referencia de las pruebas de coagulación por edad.
– National Heart, Lung and Blood Institute. The diagnosis, evaluation and management of von Willebrand Disease. Clinical Practice Guidelines. Consultado el 8/03/2016. Disponible en: http://www.nhlbi.nih.gov/health-pro/guidelines/current/von-willebrand-guidelines.
Resumen completo y actualizado de la fisiopatología, clasificación, genética, clínica, diagnóstico y tratamiento de la enfermedad de von Willebrand, con recomendaciones de manejo clínico basadas en la evidencia.
| Caso clínico |
|
Varón de 17 años, referido a consulta de Hematología Pediátrica por prolongación del tiempo de tromboblastina parcial activado (TTPA) en 2 controles preoperatorios de fimosis, con tiempo de protrombina (TP) normal. Tiene realizados estudios de coagulación a los 4 y 10 años con TTPAs normales (de 27,5 y 29,3 s) en el contexto de fiebre. Refiere en el último verano, epistaxis frecuentes por fosa nasal derecha. No otros problemas de sangrado. Antecedentes familiares: madre con equimosis fáciles desde siempre, no menorragias ni otros problemas de sangrado. Padre sano, no consanguíneo, asintomático. Un hermano de 13 años ha presentado epistaxis múltiples en el verano (sin precisar taponamiento) y tiene estudios de coagulación realizados normales. No antecedentes de enfermedades conocidas de la hemostasia. Exploración física: normal. Estudios complementarios: TP: 14,3 s. Act. protrombina: 82% (N 70-120). INR: 1,13 (N 0,8-1,2). TTPA: 36 s (N 26-35,5). Fibrinógeno: 192 mg/dL. Tiempo de obturación: colágeno/epinefrina: 143 s (N 84-160). Colágeno/ADP: 108 s (N 68-121). Prueba de mezclas (TTPA: 50% paciente: 50% control): 31,8 s (inmediato); control: 29,7 s. Incubación: 32,9 s (control: 29,7 s). Cuantificación de factores: factor de von Willebrand: Ag: 44%. Factor VIII: 55%. Factor IX: 98%, factor XI: 110%, factor XII: 86%. Test específicos para anticoagulante lúpico negativos. Hemograma: leucocitos 6.280/uL (fórmula normal). Hb: 15,5 g/dL, VCM: 87,8 fl. Plaquetas: 219.000/Ul. Centro de referencia: TP: 12,1 s. Act protrombina: 93%. INR: 1,0. TTPA: 36 s (N 26-35,5). Fibrinógeno: 184 mg/dL. Tiempo de obturación: colágeno/epinefrina: 155s (N 84-160). Colágeno/ADP: 136 s (N 68-121). Factor VIII: 47,9. Factor von Willebrand: Ag: 29,8%. Cofactor de la ristocetina: 22,9. Tras test con DDAVP: tiempo de obturación con epinefrina 60 s. Tiempo de obturación con ADP no procede. Factor VIII: 207%. Factor de von Willebrand: 123%. Cofactor de la ristocetina: 95,9 s. El día que acude a resultados al centro de referencia presentaba petequias en cuello y cara tras haber realizado ejercicios abdominales.
|


