 |
P. Mata1, R. Alonso2, J.R. Gonzalez-Juanatey3, L. Badimón4, A. Ruiz5, J.L. Díaz-Díaz6, M.T. Muñoz7, O. Muñiz8, J. Dalmau9, F. Fuentes-Jiménez10, L. Irigoyen11, E. Galve12, F.J. Ramos13, C. Sánchez14, G. Gonzalo14, J.J. Castrodeza15, J.L. Zamorano16, F. Pérez-Jiménez10
1Medicina Interna. Presidente Fundación Hipercolesterolemia Familiar, Madrid. 2Clínica de Lípidos, Medicina Interna. Fundación Jiménez Díaz, Madrid. 3Servicio de Cardiología, Hospital Universitario de Santiago de Compostela. 4Cardiovascular Research Center (CSIC-ICCC), Institut Català de Ciències Cardiovasculars, Barcelona. 5Atención Primaria. Unidad de Lípidos y Prevención Cardiovascular. Centro de Salud Pinto, Madrid. 6Medicina Interna. Hospital Abente y Lago, A Coruña. 7Servicio de Endocrinología. Hospital Infantil Universitario Niño Jesús. Departamento de Pediatría. Universidad Autónoma. Madrid. 8UCERV. UCAMI. Servicio de Medicina Interna. Hospital Virgen del Rocío. Sevilla. 9Unidad de Nutrición y Metabolopatías. Hospital Infantil La Fe, Valencia. 10IMIBIC/Hospital Universitario Reina Sofía. Universidad de Córdoba. 11Unidad de Lípidos. Servicio de Endocrinología y Nutrición. Hospital Universitario Araba, Vitoria-Gasteiz. 12Unitat d’Insuficiència Cardiaca. Servei de Cardiologia. Area del Cor. Hospital Vall d’Hebron, Barcelona. 13Presidente de la Asociación Española de Genética Humana. Especialista en Pediatría y Genética Clínica. Hospital Clínico Universitario Lozano Blesa, Zaragoza. 14Paciente, Fundación Hipercolesterolemia Familiar, Madrid. 15Ex Director Salud Pública. Consejería de Sanidad, Castilla y León. 16Servicio de Cardiología, Hospital Ramón y Cajal, Madrid. / Coordinador: Pedro Mata / Comité de redacción: Pedro Mata, Rodrigo Alonso, Jose Ramón Gonzalez-Juanatey, Lina Badimón, Antonio Ruiz. / Asesor Internacional: Gerald F. Watts. Winthrop Professor of Cardiometabolic Medicine, University of Western Australia, Australia. / Organización Promotora: Fundación Hipercolesterolemia Familiar.
Parte de este Documento de Consenso se ha publicado en Aten Primaria 2015;47(1):56-65. http://dx.doi.org/10.1016/j.aprim.2013.12.015
| Resumen
La hipercolesterolemia familiar (HF) es un trastorno genético frecuente que se manifiesta desde el nacimiento y que causa un aumento en los niveles plasmáticos de colesterol-LDL (c-LDL), xantomas y enfermedad coronaria prematura. Su detección y tratamiento precoz reduce la morbimortalidad coronaria. A pesar de la disponibilidad de un tratamiento eficaz, la HF está poco diagnosticada y tratada. La identificación de los casos índices y la posterior detección en cascada familiar, utilizando los niveles de c-LDL y la detección genética, es la estrategia más coste-efectiva para la detección de nuevos casos. El tratamiento crónico con estatinas ha disminuido el riesgo cardiovascular a los niveles de la población general. Los objetivos en c-LDL son <130 mg/dL en los niños y adultos jóvenes, <100 mg/dL en los adultos y <70 mg/dL en los adultos con enfermedad coronaria conocida o diabetes. En la mayoría de los pacientes es difícil conseguir estos objetivos, por lo que puede ser necesario el tratamiento combinado con ezetimiba u otros fármacos. Cuando no se alcanzan los objetivos con el máximo tratamiento farmacológico tolerado, una reducción de c-LDL ≥ 50% puede ser aceptable. La LDL-aféresis es útil en los pacientes homocigotos y en los heterocigotos graves resistentes al tratamiento. Este documento proporciona recomendaciones para el diagnóstico, cribado y tratamiento de la HF en niños y adultos, así como consejos específicos para los especialistas clínicos y médicos de Atención Primaria con el objetivo de mejorar el cuidado de los pacientes y reducir su carga de enfermedad cardiovascular. |
| Abstract
Familial hypercholesterolemia (FH) is a frequent genetic disorder clinically manifested since birth, associated with very high levels of plasma LDL-cholesterol (LDL-c), xanthomas and premature coronary heart disease. |
Palabras clave: Hipercolesterolemia familiar; Niños-adolescentes; Diagnóstico genético; Cribado en cascada; Enfermedad cardiovascular; Hipercolesterolemia familiar homocigota; Tratamiento
Key words: Familial hypercholesterolemia; Children-adolescents; Genetic testing; Cascade screening; Cardiovascular disease; Homozygous familial hypercholesterolemia; Treatment
Pediatr Integral 2015; XIX(7): 509.e1-509.e11
Diagnóstico y tratamiento de la hipercolesterolemia familiar en España: documento de consenso
Introducción
La hipercolesterolemia familiar (HF) es el trastorno genético más frecuente asociado con enfermedad coronaria prematura (ECP), debido a elevadas concentraciones de colesterol-LDL (cLDL) desde el nacimiento(1,2). Su mecanismo de transmisión es autosómico dominante y aproximadamente la mitad de la descendencia de una persona afecta presentará el trastorno. Se produce principalmente por mutaciones en el gen del receptor LDL (RLDL), y menos frecuentemente por mutaciones del gen de la apolipoproteína B (APOB) y del gen Proprotein Convertase Subtilisin/kexin type 9 (PCSK9)(1). Su prevalencia es de aproximadamente 1 de cada 300-500 personas en la población general, estimándose en 100.000 los españoles que padecen este trastorno(1,3).
El diagnóstico clínico se basa en concentraciones elevadas de c-LDL, historia familiar de hipercolesterolemia, antecedentes de ECP y la presencia de xantomas y/o arco corneal(1). La HF acelera la enfermedad ateroesclerótica coronaria de una a cuatro décadas(4). En España, el 55% de los varones y el 24% de las mujeres en la década de los 50 años han presentado manifestaciones de enfermedad coronaria(5). La prevalencia y el elevado riesgo de desarrollar ECP hacen de la HF un problema de salud pública. A pesar del elevado riesgo cardiovascular (RCV), la mayoría de los pacientes están sin diagnosticar ni tratar. Su diagnóstico precoz permite utilizar medidas preventivas. Entre ellas, el tratamiento crónico con estatinas ha demostrado en los pacientes con HF sin enfermedad coronaria previa una marcada reducción del RCV, similar al de la población general(6).
Necesidad de un documento de consenso para la detección y tratamiento de la hipercolesterolemia familiar
Los pacientes con HF suelen acudir al primer nivel asistencial; sin embargo, la mayoría están sin diagnosticar y por tanto sin tratamiento, o bien con tratamiento insuficiente(7,8).
En España, algunas comunidades autónomas han implementado estrategias diferentes para el diagnóstico de la HF mediante criterios clínicos y confirmación genética(9). Un estudio observacional español, con participación de médicos de Atención Primaria (AP) y Especializada, ha demostrado que menos del 5% de los casos con diagnóstico genético de HF consiguen el objetico en cLDL < 100 mg/dL y menos del 15% de estos están recibiendo el máximo tratamiento combinado(8).
Para evitar este vacío en la prevención de la enfermedad coronaria en esta población de alto riesgo, se ha elaborado este documento de consenso, cuyo objetivo es revisar la información actualmente disponible acerca del diagnóstico y tratamiento de la HF, y consensuar con un grupo de expertos, recomendaciones que ayuden a los especialistas clínicos y médicos de AP a realizar un adecuado diagnóstico, tratamiento y cribado familiar con el fin de prevenir el desarrollo de la ECP. Además, este documento puede ayudar en la elaboración de políticas de prevención y promoción de la salud.
Metodología
Para la elaboración de este documento se han seguido las recomendaciones del protocolo Appraisal of Guidelines Research & Evaluation (AGREE)(10). La Fundación Hipercolesterolemia Familiar promovió la creación de un panel de expertos formado por 6 internistas de clínicas de lípidos, 3 cardiólogos, 2 pediatras, un médico de AP, un endocrinólogo, una investigadora en ateroesclerosis, un genetista clínico, un responsable en salud y dos personas con HF. Se realizó una búsqueda en las bases Medline, PubMed y Cochrane de todos los temas relacionados con la HF.
Se realizaron dos reuniones de trabajo presenciales y dos videoconferencias en el primer semestre del 2013. Después de la revisión sistematizada de la evidencia, se discutieron y consensuaron las recomendaciones basadas en la mejor evidencia disponible, en la opinión de expertos y en la buena práctica clínica. Las recomendaciones finales se han clasificado de acuerdo a los criterios modificados del National Health and Medical Research Council(11) en: a) es de confianza para guiar la práctica clínica; b) puede ser de confianza para guiar la práctica clínica en la mayoría de las situaciones; c) puede ser utilizada para guiar la práctica clínica, pero se debe tener precaución en su aplicación.
Diagnóstico clínico en adultos
El diagnóstico de la HF se basa en niveles elevados de c-LDL (generalmente >220 mg/dL), historia familiar de hipercolesterolemia (especialmente si hay niños o adolescentes afectos), presencia de ECP y depósitos de colesterol en forma de xantomas y/o arco corneal (Tabla I y Fig. 1). Los xantomas tendinosos son patognomónicos de HF; sin embargo, se encuentran en menos del 30% de los casos confirmados de HF(8,12). Su ausencia no excluye el diagnóstico de HF.

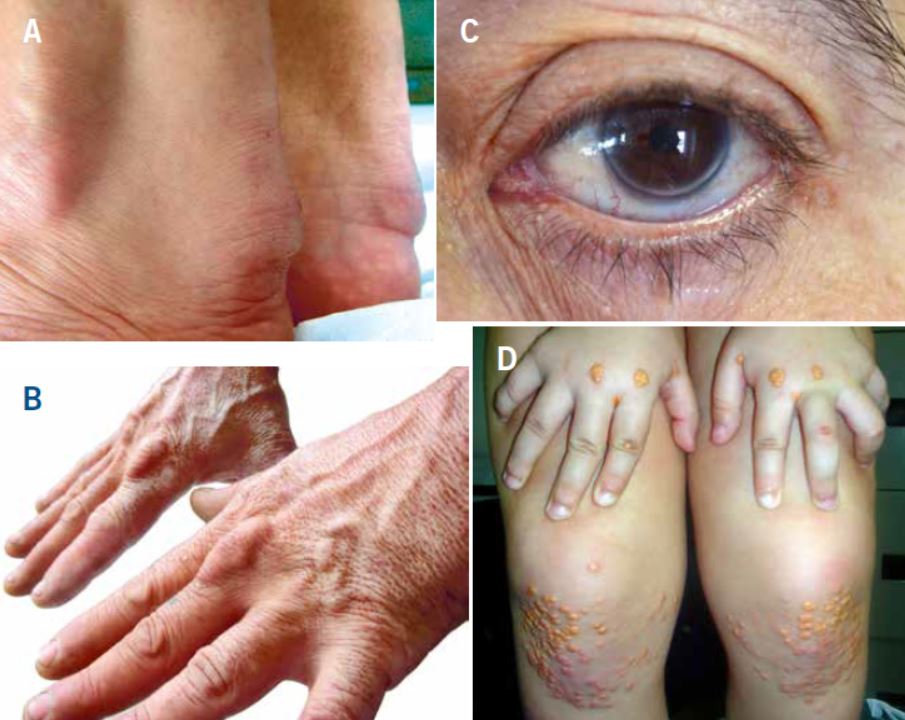
Figura 1. Signos de hipercolesterolemia familiar. A y B. Xantomas en el tendón de Aquiles y extensores de la mano; C. Arco corneal completo en un varón <45 años; D. Xantomas eruptivos y planos en manos y rodillas de un niño de 5 años con HFHo. (Fotos cortesía de la Fundación Hipercolesterolemia Familiar). Reproducido de Expert Review of Cardiovascular Therapy, March 2013, Vol. 11, No. 3, Pages 327-342, con permiso de Expert Reviews Ltd.
La obtención del árbol familiar es esencial para evaluar la probabilidad de HF y para posteriormente realizar la detección familiar. Existen tres herramientas diferentes para establecer el diagnóstico clínico de la HF en el caso índice (CI): el programa MedPed(13), el Simon Broome británico(14) y los criterios de la red de clínicas de lípidos holandesa (RCLH)(15), siendo estos últimos los que se utilizan en España. Se basan en un sistema de puntuación, según la historia personal y familiar de determinadas variables (Tabla II). El diagnóstico clínico es de certeza cuando la puntuación es ≥8 y de probabilidad cuando es ≥6. La precisión de los criterios clínicos se ha comparado con el diagnóstico genético que es el “gold standard”, siendo los criterios de la RCLH los que en conjunto tienen mejor sensibilidad y especificidad(16,17). Los criterios diagnósticos de la RCLH solo se deben utilizar para el diagnóstico del CI mayor de 18 años y nunca en sus familiares(15).

Los niveles plasmáticos elevados de triglicéridos no excluyen el diagnóstico de HF cuando la historia familiar lo apoya. El diagnóstico diferencial de la HF se debe realizar con la hiperlipidemia familiar combinada (Tabla III), la hipercolesterolemia poligénica con agregación familiar y otras causas de hipercolesterolemia secundaria como: el hipotiroidismo, el síndrome nefrótico, la colestasis y el tratamiento con determinados fármacos (esteroides, inmunosupresores, etc.).

Diagnóstico en niños y adolescentes
El diagnóstico se puede sospechar en presencia de niveles de cLDL > 190 mg/dL o bien con niveles de cLDL > 150 mg/dL cuando se tiene la confirmación genética de HF o, al menos, la evidencia de transmisión vertical de la hipercolesterolemia y/o ECP en uno de los progenitores. Se ha demostrado que los niveles de colesterol total y cLDL discriminan bien entre aquellos niños con y sin HF antes de los 10 años(18). Para el diagnóstico, se recomienda obtener la media de 2 determinaciones del perfil lipídico (preferiblemente en ayunas) con, al menos, 2 meses de diferencia debido a la variabilidad biológica en la edad infantil y descartar las causas más frecuentes de hipercolesterolemia secundaria en la infancia-adolescencia.
No hay un criterio único respecto a la edad en la que se debe hacer el diagnóstico de HF. En general, se recomienda el diagnóstico entre los 2 y los 10 años(13,14,19-21). Su importancia es que cuanto antes se realice, más fácil será la adherencia a los hábitos de vida saludables. Este panel recomienda que el diagnóstico se debe realizar a partir de los 2 años, especialmente cuando uno de los progenitores ya está diagnosticado, y a ser posible antes de los 8 años.
Diagnóstico de la hipercolesterolemia familiar homocigota
La hipercolesterolemia familiar homocigota (HFHo) es una forma rara de HF que se produce cuando se hereda la misma mutación en el gen del RLDL de ambos progenitores. Cuando se heredan mutaciones distintas en ambos alelos, se produce un cuadro conocido como “HF heterocigota compuesta” con una expresión que puede ser similar a la HFHo. Las mutaciones en los genes APOB, PCSK9 y la hipercolesterolemia autosómica recesiva (HAR) también pueden producir un fenotipo similar que varía en gravedad(1,22). Se estima que afecta a 1 caso por cada 800.000 a un millón de personas, aunque es mayor en determinadas regiones o países, presumiblemente debido a un efecto fundador y al aislamiento de una población. Los pacientes presentan hipercolesterolemia grave, xantomas y ateroesclerosis acelerada.
El diagnóstico de HFHo se debe realizar alrededor de los 2 años o inclusive antes y se basa en: concentración de cLDL sin tratamiento >500 mg/dL o cLDL con tratamiento >300 mg/dL, presencia de xantomas antes de los 10 años (Fig. 1) e historia de hipercolesterolemia o de diagnóstico genético en ambos progenitores(22). Los xantomas interdigitales, especialmente entre los dedos pulgar e índice son patognomónicos de HFHo. Ambos padres deberían tener hipercolesterolemia y/o ser heterocigotos obligados para la misma mutación causante de HF.
Habitualmente, se desarrolla una ateroesclerosis grave y generalizada que suele manifestarse clínicamente como enfermedad coronaria en edades muy jóvenes, así como con estenosis aórtica, y que si no se tratan pueden producir la muerte antes de los 20 años. Aunque la ateroesclerosis coronaria severa es la principal causa de muerte, la estenosis aórtica también es una grave complicación y causa de mortalidad en numerosos pacientes con HFHo y a menudo requiere la sustitución de la válvula aórtica(23). Se debe realizar un examen ecocardiográfico basal en los niños y después anualmente. Si aparece regurgitación aórtica, se debería realizar un test de esfuerzo y si su resultado es anormal, estaría indicada una coronariografía(23). El AngioTAC coronario es un método no invasivo que permite la detección precoz de la ateroesclerosis coronaria y aórtica y que podría ser utilizado en los adolescentes.
Diagnóstico genético
La principal ventaja de la detección de una variante funcional con efecto patogénico en los genes descritos anteriormente es que establece el diagnóstico inequívoco de la HF(24) y facilita el cribado en cascada familiar. En España, se han descrito más de 400 mutaciones en el gen del RLDL asociadas a HF(25). El diagnóstico genético solo se debe ofrecer a los casos índices con una puntuación ≥6 según los criterios de la RCLH, ya que tienen la mayor sensibilidad y especificidad(17,26-27). Se debe realizar en un laboratorio acreditado y debe incluir la secuenciación completa para identificar mutaciones puntuales y deleciones/inserciones para el gen del RLDLy de la APOB y PCSK9.
Detección de la hipercolesterolemia familiar: cribado en cascada familiar
La detección de la HF cumple los criterios de la Organización Mundial de la Salud (OMS) para el cribado sistemático de una enfermedad(15) y es coste-efectiva para detectar nuevos casos de HF(28-30). Una estrategia sistemática es esencial para la detección de los casos índices (CI) con HF. El CI es el primer miembro de una familia en ser diagnosticado y es fundamental para iniciar el cribado familiar en cascada. Los criterios diagnósticos de la RCLH solo se deben utilizar para el diagnóstico del CI mayor de 18 años y nunca en sus familiares.
En la AP, se deben buscar los CI mediante la detección oportunista basada en la historia personal y/o familiar de hipercolesterolemia y ECP (antes de los 55 años en varones y de los 60 años en mujeres). A nivel hospitalario, los CI se deben buscar entre los pacientes menores de 60 años con enfermedad coronaria e hipercolesterolemia. Para realizar el cribado en los familiares de un CI diagnosticado de HF, se recomienda usar una combinación de niveles de cLDL y análisis genético, si se dispone de los recursos necesarios(14,26-27). Diferentes estudios han demostrado que hasta el 24% de los familiares con un colesterol inferior al percentil 90 tienen un diagnóstico genético positivo, lo que justificaría realizar el análisis genético, ya que estas personas pueden transmitir el trastorno a su descendencia y expresar la hipercolesterolemia en edades más tardías(16).
Con el fin de una mejor utilización de los recursos, se recomienda que el cribado sistemático en cascada sea coordinado por un servicio especializado y dedicado que colabore estrechamente con la AP e idealmente con una organización de pacientes(14,27). Los pacientes deben ser informados de su RCV, de la importancia de informar a sus familiares para la detección precoz, así como de realizar el estudio genético si está disponible.
Riesgo cardiovascular en la hipercolesterolemia familiar
Los pacientes con HF son considerados de alto RCV(26,27). Sin embargo, el riesgo puede variar entre individuos en función de la presencia de otros factores de RCV (FRCV), especialmente la lipoproteína(a) [Lp(a)] y el tabaco, así como la presencia de ateroesclerosis subclínica (Fig. 2)(31-34). La estratificación en niveles de riesgo ayuda al médico a individualizar la intensidad del tratamiento y permite una mejor utilización de los recursos.

Figura 2. Hipercolesterolemia familiar. Evaluación y estratificación del riesgo CV en adultos. C-HDL: colesterol transportado por lipoproteínas de alta densidad; DM tipo 2: diabetes mellitus tipo 2; ECV: enfermedad cardiovascular; ECVP: enfermedad cardiovascular prematura; FRCV: factores de riesgo cardiovascular; HTA: hipertensión arterial.
La estratificación del riesgo basada en Framingham o el SCORE no es adecuada en la HF, ya que lo infravaloran sistemáticamente(26,27), especialmente en los jóvenes. En estos, una medida del RCV a largo tiempo basada en la imagen de ateroesclerosis subclínica podría ser adecuada.
La evaluación de la aterosclerosis coronaria subclínica se puede realizar de forma no invasiva, mediante la prueba de esfuerzo ECG, ecocardiografía de estrés, gammagrafía radioisotópica, angio-resonancia y el angio-TAC coronario(35-38). La evaluación de otros territorios vasculares incluye la ecografía carotídea y la determinación del índice tobillo brazo. Se recomienda evaluar la presencia de ateroesclerosis a partir de los 30 años en varones y 40 años en mujeres, o antes, si hay FRCV. En el caso de que alguna de las pruebas de imagen muestre ateroesclerosis significativa (estenosis > 50%) o una de las otras pruebas sea positiva, el paciente debe ser evaluado por Cardiología o Cirugía Vascular.
Las Guías Internacionales en HF del 2004 estratificaron el RCV en alto, intermedio y bajo(35). Sin embargo, la exposición desde el nacimiento a elevados niveles de colesterol confiere a los pacientes un alto riesgo de desarrollar una ECV prematura. Por otra parte, la presencia de un grupo de bajo riesgo dentro de un trastorno que globalmente se considera de alto riesgo puede inducir a confusión. Por tanto, este panel de expertos recomienda clasificar aquellos pacientes con HFHo, HF con enfermedad coronaria o con diabetes mellitus tipo 2 (DM2), o aquellos con evidencia de enfermedad ateroesclerótica subclínica significativa como de muy alto RCV. Los pacientes con, al menos, un FRCV deben considerarse de alto RCV y el resto de pacientes, especialmente los jóvenes, se podría considerar de RCV moderado (Fig. 2).
Objetivo terapéutico y tratamiento en adultos
Este documento recomienda un objetivo en cLDL plasmático <100 mg/dL en adultos con, al menos, un FRCV y <70 mg/dL si existe enfermedad coronaria, DM2 o la presencia de enfermedad ateroesclerótica subclínica significativa. En el resto de los pacientes (varones < 30 años y mujeres < 40 años) que no tienen otro FRCV, un c-LDL < 130 mg/dL se podría considerar aceptable (Fig. 3). Sin embargo, debido a la dificultad de conseguir este objetivo en la mayoría de los pacientes(7,8), una reducción del c-LDL ≥ 50% puede considerarse como un objetivo secundario más realista.

Figura 3. Algoritmo terapéutico en adultos con HF. cLDL: colesterol transportado por las lipoproteínas de baja densidad; FRCV: factores de riesgo cardiovascular.
La prevención de la ECV en los pacientes con HF requiere de un manejo integral en el control de los FRCV. Se debe recomendar una dieta baja en grasas saturadas, grasa trans y colesterol, así como las medidas de actividad física encaminadas a controlar el peso corporal. Tanto en niños como en adultos, se pueden utilizar alimentos enriquecidos en estanoles/esteroles vegetales que pueden reducir un 10% el c-LDL(39)>. Se debe promover el no fumar y facilitar la deshabituación en el fumador.
Todo adulto con HF debe ser tratado con medidas dietéticas y con fármacos hipolipemiantes desde el momento del diagnóstico. Varios estudios han demostrado que el tratamiento hipolipemiante intensivo tiene efectos beneficiosos, como la reducción del grosor íntima-media carotídeo y en la mejora de la función endotelial(40,41). Además, estudios observacionales han confirmado el beneficio cardiovascular de las estatinas en los pacientes con HF(6,42). La base del tratamiento es el uso de una estatina potente, como atorvastatina o rosuvastatina, en monoterapia o en combinación con ezetimiba y/o resinas sino se consigue el objetivo terapéutico. El tratamiento en adultos debe iniciarse una vez diagnosticado el trastorno y no debe interrumpirse salvo presencia de eventos adversos clínicamente relevantes o intolerancia a los fármacos. Recomendamos que la mayoría de estos pacientes sean seguidos en AP, y los casos complejos, en centros especializados.
En el caso del tratamiento de mujeres en edad fértil, se les debe informar que no hay contraindicación ni interacción con el uso de anticonceptivos orales y que, si desean quedarse embarazadas, el tratamiento debe interrumpirse desde, al menos, 2 meses antes de la concepción hasta el final del embarazo debido al riesgo potencial de anomalías fetales. Durante el embarazo y la lactancia, solo se podrían tomar las resinas.
Seguimiento y manejo de los efectos adversos de la medicación
Una vez comenzado el tratamiento, el paciente debe hacerse un control analítico a las 6-8 semanas para evaluar la respuesta, adherencia, seguridad y tolerancia al fármaco. Posteriormente, el paciente con HF debería ser visto al menos dos veces al año y en cada revisión se preguntará por los FRCV y síntomas de enfermedad cardiovascular.
Todos los tratamientos hipolipemiantes pueden inducir toxicidad hepática y muscular. Por esto, se deben obtener determinaciones plasmáticas de función hepática antes del inicio del tratamiento. Si las transaminasas basales están elevadas, se deberá realizar una ecografía hepática. El tratamiento debe interrumpirse si las transaminasas se elevan, al menos, 3 veces por encima del límite superior del valor normal en dos ocasiones en el periodo de un mes. El tratamiento puede ser reevaluado y comenzar con una estatina diferente, monitorizando las transaminasas. Los pacientes deberán informar de cualquier nueva medicación prescrita para minimizar el riesgo de interacciones farmacológicas.
Las mialgias relacionadas con las estatinas son un problema médico relevante, son dosis dependiente y varían según la estatina utilizada. En ocasiones, se puede producir una elevación asintomática de la creatinquinasa. Se debe informar a los pacientes sobre los signos de miopatía y rabdomiolisis. La mayor edad, una masa muscular reducida, la insuficiencia renal, la disfunción tiroidea y la interacción con determinados fármacos como antibióticos macrólidos o inmunosupresores como la ciclosporina, entre otros, pueden aumentar el riesgo de miopatía. Cuando se sospecha miopatía por estatinas, se debe realizar un examen físico y determinar los niveles plasmáticos de creatinquinasa. En el caso de presentar síntomas musculares graves con una estatina, debe intentarse el tratamiento con otra estatina, comenzando con dosis bajas y aumentándolas progresivamente. En estos casos, la combinación con ezetimiba puede resultar de utilidad si el paciente no tolera dosis altas de estatina.
Objetivo terapéutico y tratamiento en niños y adolescentes
Existe acuerdo en que los objetivos de tratamiento en cLDL en los niños no necesitan ser tan bajos como en los adultos y no existe evidencia para un objetivo absoluto o relativo(19,20). Este panel recomienda un cLDL plasmático < 130 mg/dL a partir de los 14 años y <160 mg/dL en los menores de 14 años, excepto si hay otro FRCV o antecedentes de enfermedad coronaria muy prematura en el progenitor afecto, en que los objetivos pueden ser más estrictos (Fig. 4).
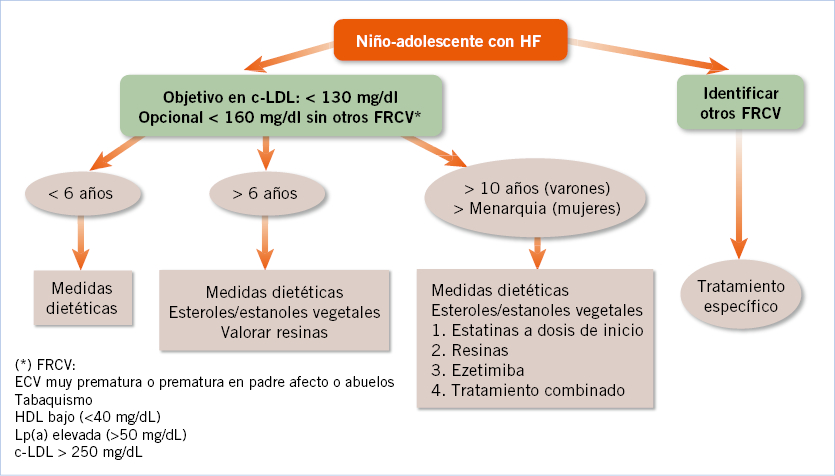
Figura 4. Algoritmo terapéutico en niños y adolescentes con HF. cLDL: colesterol transportado por las lipoproteínas de baja densidad; FRCV: factores de riesgo cardiovascular.
La alimentación es la base del tratamiento de la HF en niños y adolescentes, consiguiéndose reducciones en cLDL de hasta un 15%(21,43). Es esencial un aporte adecuado de energía y nutrientes para mantener un adecuado crecimiento y peso corporal (Tabla III). Además de una correcta alimentación, debe promoverse la actividad física, así como no fumar.
No existe unanimidad sobre a qué edad se debe comenzar el tratamiento con estatinas en niños con HF y no hay datos de seguridad antes de los 8 años(44). En los heterocigotos, se recomienda el uso de estatinas a partir de los 10 años en niños y preferiblemente después de la menarquia en las niñas si después de las medidas dietéticas el cLDL es ≥190 mg/dL, o >160 mg/dL con historia de enfermedad cardiovascular prematura en familiares de primer grado o presencia de otros FRCV(19-21). En los niños homocigotos, la medicación debe iniciarse en el momento del diagnóstico. Las estatinas son seguras y eficaces en la población infantil. Se puede utilizar cualquier estatina aprobada por las agencias regulatorias titulando la dosis según la respuesta clínica y la edad. Si con las estatinas no se consiguen los objetivos en cLDL, se debe valorar el añadir resinas o ezetimiba. Las estatinas están contraindicadas en el embarazo, por lo que debe advertirse a las adolescentes.
En todo niño en tratamiento con fármacos, debe vigilarse el ritmo de crecimiento, el desarrollo puberal y monitorizar los niveles lipídicos, transaminasas y creatinquinasa antes del inicio del tratamiento y después cada 3-6 meses(21). Los pacientes pediátricos con HF pueden ser seguidos por el pediatra, pero los pacientes que tienen un aumento grave del cLDL, numerosos FRCV o HFHo, deberían ser controlados por un especialista.
LDL-aféresis y trasplante hepático
La LDL aféresis es el tratamiento de elección para los pacientes con HFHo y ha demostrado tener un efecto beneficioso en la ateroesclerosis aórtica y coronaria(45)mejorando la supervivencia. Es una estrategia terapéutica segura y eficaz, estando descritos casos de HFHo con tratamientos de más de 20 años de duración, en los que se ha demostrado la eliminación de xantomas, la regresión angiográfica de la aterosclerosis coronaria y la reducción de los episodios coronarios mortales y no mortales(46,47). La LDL-aféresis se puede comenzar a partir de los 6 años y siempre antes de los 10 años por el elevado riesgo de estenosis aórtica grave.
Permite la eliminación específica de cLDL y Lp (a), con una disminución plasmática del 50-75% cuando se usa de forma semanal o cada 2 semanas. El tratamiento con estatinas se debe mantener para retrasar el efecto rebote en el aumento del cLDL.
Con la información disponible, se puede recomendar su indicación en las siguientes situaciones: a) HFHo a partir de los 6 años y siempre antes de los 10 años; b) HF con enfermedad coronaria sintomática y cLDL > 200 mg/dL, a pesar de tratamiento farmacológico intenso; y c) HF con enfermedad coronaria progresiva sin posibilidades de revascularización y cLDL > 125 mg/dL y Lp(a) > 60 mg/dL, a pesar de tratamiento farmacológico intenso(45,48,49). También, podría estar indicada en mujeres embarazadas con HF y ECV durante el tiempo de suspensión del tratamiento farmacológico, incluyendo la lactancia.
El trasplante de hígado puede ser una medida excepcional para los pacientes con HFHo que no son tributarios para la LDL-aféresis o que son refractarios al tratamiento farmacológico. El trasplante hepático se debe discutir con pacientes y familiares para aclarar los riesgos y beneficios del procedimiento. Antes del trasplante hepático, se debe considerar la cirugía de revascularización coronaria y el reemplazamiento de la válvula aórtica.
Papel de las organizaciones de pacientes en la detección de la hipercolesterolemia familiar y en el apoyo familiar
Se estima que en España hay unas 100.000 personas con HF, y la esperanza de vida de estos pacientes puede verse reducida de 20 a 30 años, lo que se traduciría en la pérdida potencial de 2 millones de años de vida. Además, el paciente con HF tiene una buena calidad de vida, que empeora con la presencia de enfermedad coronaria(50).
Una organización de pacientes con HF desempeña una importante función en el apoyo a los pacientes y sus familias, promoviendo el conocimiento de la HF en la comunidad, los médicos y sistemas de salud. Esta organización proporciona una red para las familias y les informa y aconseja, sobre todo lo referente a la HF. También, promueve y desarrolla servicios dentro de la comunidad, facilita el aprendizaje y comparte información que va ser fundamental para la detección, el manejo y la reducción del riesgo cardiovascular. El paciente informado toma parte activa en el cuidado de su salud, hace un uso más racional de los medicamentos y cumple mejor el tratamiento crónico. Además, una organización de pacientes puede proporcionar los medios para establecer un registro tanto de pacientes como de los servicios disponibles, incluyendo los centros que realizan el cribado en cascada para la detección de la HF.
En 1997, y de acuerdo con las recomendaciones de la OMS, se crea en España la Fundación de Hipercolesterolemia Familiar (FHF), organización benéfica asistencial sin ánimo de lucro (www.colesterolfamiliar.org). Su misión es informar, educar, detectar y apoyar a las familias con HF. Entre sus logros, destaca la obtención de la aportación reducida al tratamiento crónico con estatinas y ezetimiba.
En los últimos años, algunas comunidades autónomas están realizando el diagnóstico genético de HF financiado por el sistema de salud, y en la actualidad hay cerca de 7.000 pacientes en España identificados genéticamente, cerca del 50% gracias al programa de detección de la FHF en colaboración con centros hospitalarios de toda España (http://safeheart.colesterolfamiliar.org/).
Programas de detección regionales
En España, las comunidades de Aragón, Asturias, Castilla y León, Cataluña, La Rioja, Madrid, Navarra y País Vasco han implementado diferentes estrategias de detección de la HF, incluyendo el diagnóstico genético.
Castilla y León ha sido la única comunidad que en su estrategia de detección ha incluido a los médicos de AP (http://www.saludcastillayleon.es). Esta comunidad, en colaboración con la FHF viene desarrollando desde 2009 el Programa de detección precoz de la HF, que incluye la formación de los médicos y la creación de un registro ad hoc. El médico de AP o de Atención Especializada selecciona el CI siguiendo los criterios diagnósticos de la RCLH (puntuación ≥ 6). Una vez identificado, se solicita el estudio genético en muestra de saliva. Si se confirma el diagnóstico, se procede a realizar la detección en cascada familiar a los familiares de primer grado, sin necesidad de aplicar los criterios clínicos de la RCLH. Hasta la fecha, se han diagnosticado genéticamente unos 1.000 pacientes con HF. Este programa debería servir de modelo para el resto de comunidades que todavía no lo están realizando y para impulsar una estrategia homogénea a nivel nacional.
Resumen de recomendaciones
1. Diagnóstico de HF en adultos:
1.1 Se debe sospechar una HF en un adulto con cLDL > 220 mg/dL y antecedentes de hipercolesterolemia y/o ECP en familiares de primer grado. (A)
1.2 Los criterios de la RCLH se deben utilizar solamente para el diagnóstico del caso índice (CI) y siempre en > 18 años. (A)
1.3 Para detectar a los CI se debe realizar una búsqueda oportunista en los pacientes con ECP y en aquellos con antecedentes personales y/o familiares de hipercolesterolemia y/o ECP. (A)
2. Diagnóstico de HF en niños y adolescentes:
2.1 Se debe sospechar una HF en niños con cLDL ≥ 190 mg/dL o cLDL > 150 mg/dL con historia de hipercolesterolemia y/o ECP en uno de los progenitores. (A)
2.2 Se debería realizar la determinación de colesterol en los niños a partir de los 2 años si existe historia de HF en uno de los progenitores. (A)
2.3 Si se conoce el defecto genético en uno de los padres, se puede realizar el diagnóstico genético en el niño con cLDL > 150 mg/dL, previa información y consentimiento del progenitor afectado o tutor legal. (A)
3. Diagnóstico de HF Homocigota:
3.1 Se debe sospechar en presencia de un cLDL > 500 mg/dL sin tratamiento. (A)
3.2 Se debe sospechar en presencia de un cLDL > 300 mg/dL con el máximo tratamiento farmacológico. (B)
3.3 La presencia de xantomas tuberosos o tendinosos antes de los 10 años de edad sugiere el diagnóstico de HFHo. (A)
4. Diagnóstico genético de la HF:
4.1 Se recomienda realizar el diagnóstico genético de HF en un CI con una puntuación ≥ 6 puntos de acuerdo a criterios clínicos de la RCLH. (A)
4.2 Cuando se detecta una mutación en el CI se recomienda el cribado genético en cascada familiar. (A)
4.3 La no detección de una mutación no excluye el diagnóstico de HF cuando el fenotipo es sugerente. (B)
5. Cribado familiar:
5.1 El cribado en cascada familiar debería combinar niveles de colesterol total y cLDL y análisis genético si está disponible. (A)
5.2 El cribado en cascada familiar debería estar centralizado y requiere la coordinación entre los médicos de AP y de Atención Especializada, así como de enfermería. (A)
6. Riesgo cardiovascular en la HF:
6.1 Los pacientes con HF se deben considerar de alto RCV. Sin embargo, en algunos casos se podría estratificar el riesgo. (A)
6.2 Los pacientes con HF y con, al menos, un FRCV tienen un RCV alto. (A)
6.3 Los pacientes con HFHo y con HF que presentan ateroesclerosis subclínica significativa, ECV clínica o DM2 tienen un RCV muy alto. (A)
6.4 Se debe medir los niveles de Lp(a) en todos los pacientes con HF. (A)
6.5 En pacientes con HF heterocigota > 30 años (varones) y > 40 años (mujeres) sin clínica de ECV se debería realizar una búsqueda de enfermedad aterosclerótica mediante técnicas no invasivas, aunque su utilidad no se ha validado en la HF. (C)
7. Objetivo y tratamiento de la HF en adultos
7.1 En los pacientes con HF y con RCV muy alto, el objetivo de cLDL debería ser <70 mg/dL. (A)
7.2 En los pacientes con HF y riesgo CV alto, el objetivo de cLDL debería ser <100 mg/dL. (A)
7.3 En el resto de los pacientes adultos con HF, se podría considerar un cLDL < 130 mg/dL. (B)
7.4 En el caso de no alcanzar el objetivo en cLDL con el máximo tratamiento tolerado, se debe conseguir, al menos, una reducción de cLDL > 50%. (A)
7.5 Se recomienda la administración de estatinas potentes a dosis altas y, si es necesario, en combinación con ezetimiba y/o resinas. (A)
8. Objetivo y tratamiento de la HF en niños y adolescentes
8.1 Se puede comenzar tratamiento con estatinas a partir de los 10 años en los varones y preferiblemente después de la menarquia en las niñas. (A)
8.2 El objetivo en cLDL debería ser <130 mg/dL y se podría considerar un cLDL < 160 mg/dL en los < 14 años sin otro FRCV. (B)
8.3 Es necesario monitorizar el crecimiento y desarrollo puberal, así como los niveles de transaminasas y creatinquinasa al inicio, a los 3 meses de tratamiento y posteriormente de forma anual. (A)
8.4 Los niños con HFHo deben iniciar el tratamiento farmacológico en el momento del diagnóstico e idealmente no más tarde de los 2 años. (A)
9. LDL-aféresis y otras terapias:
9.1 La LDL-aféresis se puede realizar en los pacientes con HFHo a partir de los 6 años y en casos seleccionados de HF heterocigota grave. (A)
9.2 El efecto de la LDL-aféresis en la progresión de la ateroesclerosis se debe monitorizar con ecocardiografía de la válvula y raíz aórtica, ecografía de carótida y test de esfuerzo. (B)
9.3 El trasplante hepático puede ser una alternativa excepcional en los casos de HFHo refractarios al tratamiento. (C)
Agradecimientos
Agradecemos al Dr. Gerald Watts (Universidad de Perth, Australia) por la lectura del documento y por sus constructivos y expertos consejos. A los Dres.: Rosa de los Ríos, Guillermo Domenech, Milagros Joral e Isabel Alonso de la Dirección General de Salud Pública de la consejería de Castilla y León, por su contribución al programa de detección de la HF en esa Comunidad. A María Teresa Pariente de la FHF, por su dedicación en la coordinación del programa de detección en cascada familiar de la HF, y a las familias por su participación.
Financiación
Este trabajo ha sido apoyado por la Fundación Hipercolesterolemia Familiar. El programa de detección genética de la HF en cascada familiar ha contado con fondos de la Red de Hiperlipemias Genéticas del Instituto de Salud Carlos III (ISCIII) G03/181, Centro de Investigación Cardiovascular (CNIC) 08-2008 y FIS PI12/01289 del ISCIII.
Bibliografía
1. Goldstein JL, Hobbs HH, Brown MS. Familial hypercholesterolemia. In: The Metabolic and Molecular Basis of Inherited Disease (Volume II). Scriver CR, Beaudet AL, Sly WS, Valle D (Eds). McGraw-Hill, NY, USA, 2863–2913 (2001).
2. Slack J. Risks of ischaemic heart-disease in familial hyperlipoproteinaemic states. Lancet. 1969; 2: 1380-2.
3. Benn M, Watts GF, Tybjaerg-Hansen A, Nordestgaard BG. Familial Hypercholesterolemia in the Danish General Population: Prevalence, Coronary artery disease, and cholesterol-lowering medication. J Clin Endocrinol Metab. 2012; 97: 3956-64.
4. World Health Organization. Familial Hypercholesterolaemia: Report of a WHO consultation. Paris: World Health Organisation; 1997.
5. Alonso R, Castillo S, Civeira F, Puzo J, de la Cruz JJ, Pocovi M, Mata P. Heterozygous familial hypercholesterolemia in Spain. Description of 819 non related cases. Med Clin (Barc). 2002; 118: 487-92.
6. Versmissen J, Oosterveer DM, Yazdanpanah M, Defesche JC, Basart DC, Liem AH, et al. Efficacy of statins in familial hypercholesterolaemia: a long term cohort study. BMJ. 2008; 337: a2423.
7. Pijlman AH, Huijgen R, Verhagen SN, Imholz BP, Liem AH, Kastelein JJ, et al. Evaluation of cholesterol lowering treatment of patients with familial hipercholesterolemia: A large cross-sectional study in The Netherlands. Atherosclerosis. 2010; 209: 189-94.
8. Mata N, Alonso R, Badimón L, Padró T, Fuentes F, Muñiz O, et al. Clinical characteristics and evaluation of LDL-cholesterol treatment of the Spanish Familial Hypercholesterolemia Longitudinal Cohort Study (SAFEHEART). Lipids Health Dis. 2011; 10 (94).
9. Mata P, Alonso R, Pérez-Jiménez F. Detección de la Hipercolesterolemia Familiar: un modelo de medicina preventiva. Rev Esp Cardiol. 2014; 67: 65-8.
10. The AGREE Collaboration. Development and validation of an international appraisal instrument for assessing the quality of clinical practice guidelines: the AGREE project. Qual Saf Health Care. 2003; 12: 18-23.
11. National Health and Medical Research Council (NHMRC). NHMRC additional levels of evidence and grades for recommendations for developers of guidelines. Pilot program 2005-2007; 2005.
12. Descamps OS, Leysen X, Van Leuven F, Heller FR. The use of Achilles tendon ultrasonography for the diagnosis of familial hypercholesterolemia. Atherosclerosis. 2001; 157: 514-8.
13. Goldberg AC, Hopkins PN, Toth PP, Ballantyne CM, Rader DJ, Robinson JG, et al. National Lipid Association Expert Panel on Familial Hypercholesterolemia. Familial hypercholesterolemia: scree-ing, diagnosis and management of pediatric and adult patients: clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011; 5 (Suppl. 3): S1–S8.
14. Wierzbicki AS, Humphries SE, Minhas R. Guideline Development Group. Familial hypercholesterolemia: summary of NICE guidance. BMJ. 2008; 337, a1095. doi:10.1136/bmj.a1095 (Complete guideline in website: http://www.nice.org.uk/nicemedia/pdf/CG071NICEGuideline.pdf)
15. World Health Organization. Familial Hypercholesterolemia: Report of a second WHO consultation, World Health Organisation, Human Genetics programme, Division of Non-communicable Diseases. World Health Organization, Geneva, Switzerland (1999) (Who publication no. WHO7HGN/FH/ CONS/99.2).
16. Damgaard D, Larsen ML, Nissen PH, Jensen JM, Jensen HK, Soerensen VR, et al. The relationship of molecular genetic to clinical diagnosis of familial hypercholesterolemia in a Danish population. Atherosclerosis. 2005; 180: 155-60.
17. Civeira F, Ros E, Jarauta E, Plana N, Zambon D, Puzo J, et al. Comparison of genetic versus clinical diagnosis in familial hypercholesterolemia. Am J Cardiol. 2008; 102: 1187-93.
18. Wald DS, Bestwick JP, Wald NJ. Child-parent screening for familial hypercholesterolaemia: Screening strategy based on a meta-analysis. BMJ. 2007; 335: 599.
19. Descamps OS, Tenoutasse S, Stephenne X, Gies I, Beauloye V, Lebrethon MC, et al. Management of familial hypercholesterolemia in children and young adults: Consensus paper developed by a panel of lipidologists, cardiologists, paediatricians, nutritionists, gastroenterologists, general practitioners and a patient organization. Atherosclerosis. 2011; 218: 272-80.
20. Watts GF, Sullivan DR, Poplawski N, van Bockxmeer F, Hamilton-Craig I, Clifton PM, et al. Familial hypercholesterolaemia: A model of care from Australasia. Atherosclerosis. 2011; (Suppl 12): 221-63.
21. Morais A, Lama RA, Dalmau J, Comité de Nutrición de la AEP. Hipercolesterolemia. Abordaje terapéutico. An Pediatr (Barc). 2009; 70: 488-96.
22. Raal FJ, Santos R. Homozygous familial hypercholesterolemia: Current perspectives on diagnosis and treatment. Atherosclerosis. 2012; 223: 262-8.
23. Kolansky DM, Cuchel M, Clark BJ, Paridon S, McCrindle BW, Wiegers SE, et al. Longitudinal evaluation and assessment of cardiovascular disease in patients with homozygous familial hypercholesterolemia. Am J Cardiol. 2008; 102: 1438-43.
24. Huijgen R, Kindt I, Fouchier SW, Defesche JC, Hutten BA, Kastelein JJ, et al. Functionality of sequence variants in the genes coding for the low-density lipoprotein receptor and apolipoprotein B in individuals with inherited hypercholesterolemia. Hum Mutat. 2010; 31: 752-60.
25. Palacios L, Grandoso L, Cuevas N, Olano-Martín E, Martínez A, Tejedor D, et al. Molecular characterization of familial hypercholesterolemia in Spain. Atherosclerosis. 2012; 221: 137-42.
26. Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, et al. European Atherosclerosis Society Consensus Panel. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013; 34: 3478-90a. doi: 10.1093/eurheartj/eht273.
27. Watts GF, Gidding S, Wierzbicki AS, Toth PP, Alonso R, Brown WV, et al. Integrated guidance on the care of familial hypercholesterolaemia from the International FH Foundation. Int J Cardiol. 2014; 171(3): 309-25. doi: 10.1016/j.ijcard.2013.11.025.
28. Ned RM, Sijbrands EJ. Cascade Screening for Familial Hypercholesterolemia (FH). PLoS Curr. 2011 May. 23; 3: RRN1238.
29. Oliva, J, López-Bastida J, Moreno SG, Mata P, Alonso R. Cost-effectiveness analysis of a genetic screening program in the close relatives of Spanish patients with familial hypercholesterolemia. Rev Esp Cardiol. 2009; 62: 57-65.
30. Alonso R, Defesche J, Tejedor D, Castillo S, Stef M, Mata N, et al. Genetic diagnosis of familial hypercholesterolemia using a DNA-array based platform. Clin Biochem. 2009; 42: 899-903.
31. Alonso R, Andrés E, Mata N, Fuentes-Jiménez F, Badimón L, López-Miranda J, et al. Lipoprotein(a) levels in Familial Hipercholesterolaemia: an important predictor for cardiovascular disease independent of LDL-receptor gene mutation. J Am Coll Cardiol. 2014; 63: 1982-9.
32. Alonso R, Mata N, Castillo S, Fuentes F, Sáenz P, Muñiz O, et al. Cardiovascular disease in familial hypercholesterolaemia: Influence of low-density lipoprotein receptor mutation type and classic risk factors. Atherosclerosis. 2008; 200: 315-22.
33. Hopkins PN, Stephenson S, Wu LL, Riley WA, Xin Y, Hunt SC. Evaluation of coronary risk factors in patients with heterozygous familial hypercholesterolemia. Am J Cardiol. 2001; 87: 547-53.
34. Jansen AC, van Aalst-Cohen ES, Tanck MW, Trip MD, Lansberg PJ, Liem AH, et al. The contribution of classical risk factors to cardiovascular disease in familial hypercholesterolaemia: Data in 2400 patients. J Intern Med. 2004; 256: 482-90.
35. Civeira F. International panel on management of familial hypercholesterolemia. Guidelines for the diagnosis and management of heterozygous familial hypercholesterolemia. Atherosclerosis. 2004; 173: 55-68.
36. Alonso R, Mata P, Zambón D, Mata N, Fuentes-Jiménez F. Early diagnosis and treatment of familial hypercholesterolemia: improving patient outcomes. Expert Rev Cardiovasc Ther. 2013; 11: 327-42.
37. Miname MH, Ribeiro MS 2nd, Parga Filho J, Avila LF, Bortolotto LA, Martínez LR, et al. Evaluation of subclinical atherosclerosis by computed tomography coronary angiography and its association with risk factors in familial hypercholesterolemia. Atherosclerosis. 2010; 213: 486–91.
38. Caballero P, Alonso R, Rosado P, Mata N, Fernández-Friera L, Jiménez-Borreguero LJ, et al. Detection of subclinical atherosclerosis in familial hypercholesterolemia using non-invasive imaging modalities. Atherosclerosis. 2012; 222: 468-72.
39. Musa-Veloso K, Poon TH, Elliot JA, Chung C. A comparison of the LDL-cholesterol lowering efficacy of plant stanols and plant sterols over a continuous dose range: Results of a meta-analysis of randomized, placebo-controlled trials. Prostaglandins Leukot Essent Fatty Acids. 2011; 85 :9-28.
40. Smilde TJ, van Wissen S, Wollersheim H, Trip MD, Kastelein JJ, Stalenhoef AF. Effect of aggressive versus conventional lipid lowering on atherosclerosis progression in familial hypercholesterolaemia (ASAP): A prospective, randomised, double-blind trial. Lancet. 2001; 357: 577-81.
41. Alonso R, Mata P, De Andrés R, Villacastin BP, Martínez-González J, Badimon L. Sustained long-term improvement of arterial endothelial function in heterozygous familial hypercholesterolemia patients treated with simvastatin. Atherosclerosis. 2001; 157: 423-9.
42. Neil A, Cooper J, Betteridge J, Capps N, McDowell I, Durrington P, et al. Reductions in all-cause, cancer, and coronary mortality in statin-treated patients with heterozygous familial hypercholesterolaemia: a prospective registry study. Eur Heart J. 2008; 29: 2625-33.
43. Poustie VJ, Rutherford P. Dietary treatment for familial hypercholesterolemia. Cochrane Database Syst Rev. 2001; 2: CD001918.
44. Vuorio A, Docherty KF, Humphries SE, Kuoppala J, Kovanen PT. Statin treatment of children with familial hypercholesterolemia-trying to balance incomplete evidence of long-term safety and clinical accountability: Are we approaching a consensus? Atherosclerosis. 2013; 226: 315-20.
45. Thompson GR, Catapano A, Saheb S, Atassi-Dumont M, Barbir M, Eriksson M, et al. Severe hypercholesterolaemia: Therapeutic goals and eligibility criteria for LDL apheresis in Europe. Curr Opin Lipidol. 2010; 21: 492-8.
46. Thompson GR, Maher VM, Matthews S, Kitano Y, Neuwirth C, Shortt MB, et al. Familial Hypercholesterolaemia Regression Study (LAARS): A randomised trial of low density-lipoprotein apheresis. Lancet. 1995; 345: 811-6.
47. Sachais BS, Katz J, Ross J, Rader DJ. Long-term effects of LDL apheresis in patients with severe hypercholesterolemia. J Clin Apher. 2005, 20: 252-5.
48. Szczepiorkowski ZM, Winters JL, Bandarenko N, Kim HC, Linenberger ML, Marques MB, et al. Guidelines on the use of therapeutic apheresis in clinical practice-evidence-based approach from the Apheresis Applications Committee of the American Society for Apheresis. J Clin Apher. 2010; 25: 83-177.
49. Ito MK, McGowan MP, Moriarty PM. Management of familial hypercholesterolemias in adult patients: Recommendations from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011; 5(Suppl. 3): S38-S45.
50. Mata N, Alonso R, Banegas JR, Zambón D, Brea A, Mata P. Quality of life in a cohort of familial hypercholesterolemia patients from the south of Europe. Eur J Public Health 2012, doi:10.1093/eurpub/cks174.
