Obstrucción crónica de la vía aérea superior
Introducción
Se hará referencia primero al primer y tercer tramos (fosas nasales y laringe), dejando el segundo (rinofaringe y orofaringe) para el final del artículo, debido a que, en la práctica diaria, es en el que se concentran el mayor porcentaje de las obstrucciones crónicas. También, se justifica por el hecho de que la hipertrofia amigdalar sea la responsable de la apnea obstructiva del sueño a la que, por su incidencia en el momento actual, se dedica una atención especial.
Fosas nasales
El primer tramo de la vía aérea superior lo comprenden las fosas nasales.
Su obstrucción, no por su frecuencia, deja de ser importante, ya que no tan sólo se encarga de hacer llegar el aire a los pulmones en unas condiciones óptimas de temperatura, humedad y pureza, sino que su alteración en épocas de tan intenso crecimiento puede modificar el desarrollo del macizo facial anterior.
Salvo en pocas ocasiones, los síntomas que acompañan a la obstrucción no nos ayudan al diagnóstico etiológico. La clave del diagnóstico diferencial la tiene la historia clínica. Deberemos tener en cuenta la edad del paciente, el tiempo de evolución del proceso, su posible estacionalidad, si es uni o bilateral, si existen antecedentes traumáticos o quirúrgicos, la exposición a irritantes o tóxicos ambientales, la acción de medicamentos y, en lactantes, el tratamiento al que pueda estar sometida la madre. Se considera actualmente que un niño de cinco años es capaz de describir perfectamente sus síntomas. Sus explicaciones, junto a las observaciones de padres, maestros y cuidadores, y una exploración adecuada, conseguirán un elevado porcentaje de diagnósticos correctos.
Etiología
Las causas de obstrucción crónica de las fosas nasales son numerosas. Fundamentalmente, se pueden dividir en cuatro grupos: congénitas, inflamatorias, traumáticas y tumorales.
Congénitas
1. Atresia de coanas. Es la anomalía congénita nasal más frecuente. Consiste en la impermeabilidad posterior de una o de las dos fosas nasales. La bilateral puede ser causa de urgencia vital neonatal; sin embargo, algunas unilaterales pueden pasar desapercibidas hasta la adolescencia. El hecho de no poder pasar una sonda más de 35 mm por las fosas nasales, debe hacernos sospechar su existencia. Se clasifican en óseas (29%) y osteomembranosas (71%). La TC es la exploración radiológica obligada para establecer el diagnóstico, valorar las características de la atresia y elegir la técnica quirúrgica más adecuada. Dependiendo de si es uni o bilateral, ósea u osteocartilaginosa, el tratamiento será urgente o diferido y más o menos agresivo. Desde abordajes transnasales y transantrales a abordajes transpalatinos(1,2).
2. Paladar hendido, asociado o no a fisura palatina y a labio leporino. Provoca diferentes grados de obstrucción y de dificultad de alimentación. Las podemos dividir en completas o incompletas y unilaterales o bilaterales. Las secuelas tendrán una magnitud variable, la que menos tiene es la hendidura incompleta. El tratamiento es siempre quirúrgico y laborioso(1,2).
3. Meningoceles y meningoencefaloceles. Son herniaciones extracraneales de meninges y de tejido cerebral. Dependiendo de su localización y tamaño, producen diferentes grados de obstrucción; debido a esto, algunos no son diagnosticados más que por la casualidad en edad adulta. Se hallan cubiertos por mucosa nasal, son elásticos y pulsátiles y suelen aumentar con las maniobras de Valsalva. Si bien la TC nos puede mostrar significativos defectos en la base del cráneo, la prueba diagnóstica de elección es la RM. Jamás debemos practicar una biopsia sin haber realizado antes esta prueba, ya que, hasta que no se demuestre lo contrario, una masa endonasal en la primera infancia es un meningocele. El tratamiento es quirúrgico, bien desde la nariz mediante cirugía endoscópica nasosinusal, o bien mediante complejos abordajes neuroquirúrgicos(1-3).
4. Gliomas. Son masas de tejido glial en una localización heterotópica extradural. No se trata de un tumor, simplemente se trata de tejido cerebral aislado al cerrarse el foramen caecum. Su localización intranasal (30%) produce obstrucción respiratoria, tanto si su localización es anterior como posterior. Un 50% conservan una comunicación con la duramadre. Se puede confundir con un pólipo y su diagnóstico diferencial se establece con los meningoencefaloceles. A diferencia de estos, es fijo, no es pulsátil y no se modifica con las maniobras de Valsalva. Su tratamiento es quirúrgico y exige, en ocasiones, abordajes intracraneales(1,2).
5. Quistes del dorso nasal. Son de especial importancia, ya que, por su localización, interfieren en el desarrollo de huesos y cartílagos nasales. Son, en general, visibles desde el nacimiento y se abren al exterior en la unión osteocartilaginosa del dorso. Son fácilmente diagnosticables si aparece supuración o pelos. Su exéresis completa es el único tratamiento curativo(1-3).
Inflamatorias
1. Alergia: es una causa frecuente de obstrucción nasal en el niño. Sus síntomas facilitan el diagnóstico etiológico de la obstrucción, sobre todo si es estacional. Aproximadamente, un 20% de los niños de 2 a 3 años pueden tener síntomas de rinitis alérgica, hasta un 40% a los 6 años y un 30% pueden verse afectados durante la adolescencia. Su aumento de incidencia se atribuye a diferentes causas: mayor número de animales en las casas, aditivos y colorantes alimentarios, polución ambiental, tabaco y alcohol durante el embarazo, contacto en guarderías con diferentes virus respiratorios, etc. La clínica característica de rinitis alérgica no se manifiesta plenamente hasta los cuatro o cinco años y se va incrementando hasta la adolescencia. El diagnóstico se basa en test in vivo, como son las pruebas cutáneas (PRICK) y el test de provocación nasal, y test in vitro, como la detección de IgE específica (RAST). El tratamiento tendrá tres vertientes: sintomatológico (antihistamínicos, corticoides tópicos, etc.), de evitación del alergeno y etiopatogénico, mediante la inmunoterapia(2,4).
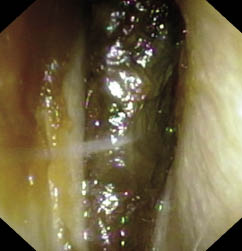
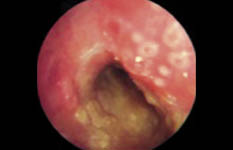
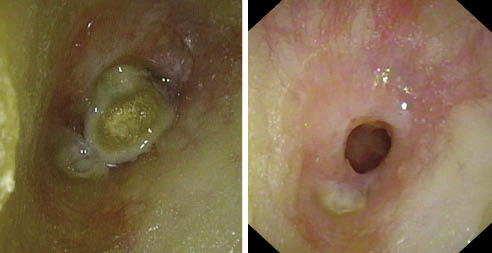
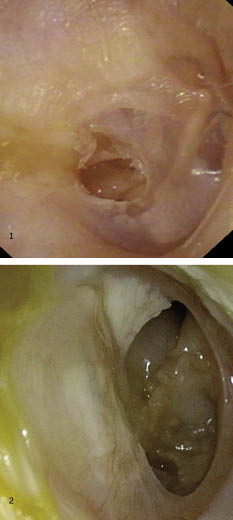
2. Rinosinusitis: la cavidad nasal y los senos paranasales constituyen en el niño una cavidad anatómica en evolución, con una estrecha dependencia, tanto en el plano fisiológico como patológico. Se acepta como rinosinusitis crónica, aquella infección sinusal de síntomas leves cuya evolución se prolonga más de cuatro semanas. A pesar de que se ha considerado durante años que el origen era infeccioso, es muy probable que el origen sea un edema de la mucosa nasal y sinusal que provoque una alteración de la ventilación, del movimiento ciliar y un enlentecimiento del transporte del moco, circunstancias que favorecerán la colonización de los senos por las cepas más frecuentes del aparato respiratorio (Streptococcus pneumoniae, Haemophilus influenzae y Moxarella catharralis, entre otras) (Fig. 1). Cualquier proceso que provoque una alteración del complejo osteomeatal (infecciones virales, alergia, irritantes, etc.) provoca una alteración de la ventilación y del drenaje de los senos que, a su vez, provoca una disminución de la presión parcial de oxígeno, una disminución de la presión dentro del seno y, como consecuencia, un enlentecimiento del transporte mucociliar y una alteración del pH, circunstancias que favorecen la proliferación de la flora. Una de las causas que pueden provocar este proceso es el reflujo gastroesofágico. De difícil diagnóstico en el niño, debe descartarse cuando nos encontremos frente a una rinosinusitis crónica o recurrente, rebelde al tratamiento(2,4-6).

Figura 1. Imagen endoscópica de una rinosinusitis purulenta.
Todas las enfermedades sistémicas que alteran la viscosidad del moco o la movilidad ciliar provocando un estasis del moco, pueden ser causa de afecciones sinusales recurrentes.
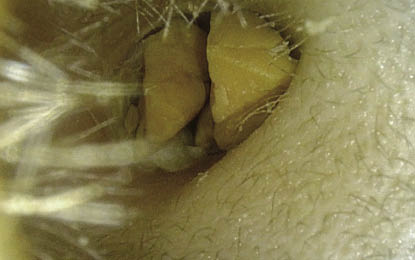
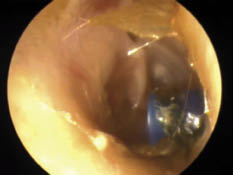
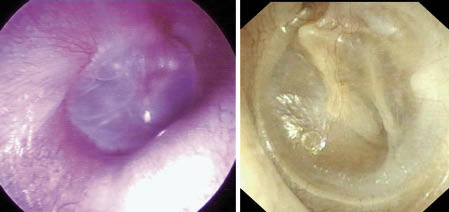
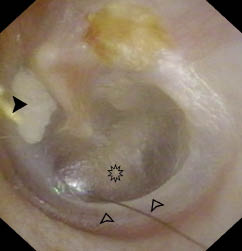
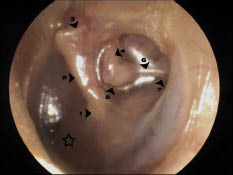
3. Poliposis nasosinusal: los pólipos son la tumoración benigna más frecuente de las fosas nasales (Fig. 2). Son neoformaciones de carácter inflamatorio que, en general, se originan en el etmoides. Pueden ser unilaterales o bilaterales. El pólipo unilateral más frecuente es el antrocoanal de Killian (Fig. 3).

Figura 2. Imagen endoscópica de una poliposis nasal (pólipo que ocupa el meato medio).

Figura 3. Imagen endoscópica de un pólipo antrocoanal de Killian a su salida del seno maxilar.
Pueden presentarse de forma aislada o asociados a asma bronquial, teniendo algunas de estas asociaciones connotaciones características, como la enfermedad de Widal o tríada ASA, en la que, además, hay una intolerancia a la aspirina.
Producen diferentes grados de obstrucción dependiendo de su tamaño y, en algunos casos, tienen tanta fuerza que, en edades tempranas, pueden deformar la pirámide nasal en su crecimiento. Es el caso de la poliposis nasosinusal infantil deformante o enfermedad de Woakes.
Las poliposis que afectan a las fosas nasales de forma bilateral pueden ser de causa alérgica o no (la mayoría), y pueden ser la expresión local de enfermedades sistémicas más graves, como la fibrosis quística(2,4).
Traumáticas
1. Los traumatismos nasales son la principal causa de desviaciones septales, sin embargo, en un gran número se deben a partos laboriosos. En la mayoría de los casos, se afecta el septum cartilaginoso, pero también se pueden fracturar los huesos propios de la nariz. Deben ser valorados con precisión y actuar en consecuencia; ya que, independientemente de la obstrucción que provoquen, pueden, en su crecimiento, alterar la morfología de la pirámide nasal.
La exploración nos ha de permitir valorar con exactitud el tipo de lesiones; ya que, de ello dependerá el tipo de tratamiento, que puede ir, desde mantener una actitud expectante, hasta realizar una cirugía de mayor o menor complejidad. Si bien, hoy en día, la cirugía septal o septopiramidal se acepta como un acto que se debe realizar para prevenir futuras complicaciones, debe ser extremadamente limitada para no alterar los centros de crecimiento(1).
2. Cuerpos extraños. A pesar de que una obstrucción nasal unilateral, sobre todo si se acompaña de rinorrea mucopurulenta y más aún si es maloliente, debe hacernos pensar inmediatamente en un cuerpo extraño, algunos de ellos son diagnosticados meses después de la obstrucción y múltiples tratamientos médicos(2).
Tumorales
1. Tumores benignos: en la infancia, la mayoría son de origen vascular.
– Hemangiomas cavernosos de septum o cornetes. Por su localización pueden provocar obstrucción; por ello, deben ser extirpados quirúrgicamente.
2. Tumores malignos: son tumores raros y, al no pensar en ellos debido a la poca especificidad de su sintomatología en estados iniciales, pueden pasar desapercibidos perdiéndose posibilidades de curación.
– El sarcoma más frecuente en la infancia es el rabdomiosarcoma.
Su tratamiento varía dependiendo de su tamaño y delimitación. Si son pequeños y bien delimitados, los extirparemos completamente con márgenes de seguridad e irradiaremos. En los grandes tumores, la única actuación quirúrgica será para hacer una biopsia. Se tratarán con quimioterapia y radioterapia asociada(2,7).
– Neuroblastoma olfatorio. Son tumores que se originan en la membrana olfatoria. Forman una lesión localmente agresiva y metastatizan en el 30% de los casos en ganglios regionales, pulmón y huesos. La supervivencia es elevada y su pronóstico depende de si sólo afecta a las fosas nasales, se extiende a los senos o de si lo hace más allá de la cavidad nasosinusal.
El tratamiento de elección es la cirugía radical seguida de radioterapia.
Laringe
El tercer tramo de este estudio lo forma la laringe.
La laringe del niño no es simplemente una miniatura de la del adulto, sino que, en cuanto a sus funciones y conformación, existen importantes diferencias. Los cartílagos y el resto de las estructuras laríngeas son mucho más flexibles y maleables y, por lo tanto, son más susceptibles al colapso y menos resistentes al edema(6).
La obstrucción laríngea puede ser congénita o adquirida (Tabla I).

Obstrucción post-traqueotomía
La traqueotomía en los niños tiene una morbi-mortalidad mayor que en los adultos. Una técnica quirúrgica incorrecta es la causa de muchas complicaciones y secuelas, en concreto de estenosis. Por ello, es recomendable que se realice siempre por manos expertas y con una técnica quirúrgica minuciosa(8).
Obstrucción post-intubación prolongada
La intubación prolongada puede producir la aparición de: granulomas, localizados preferentemente en el tercio posterior de las cuerdas vocales, quistes subglóticos o estenosis laringotraqueales, que aparecen en el 1-8% de los casos. La endoscopia es, sin duda, la prueba diagnóstica de elección para esta patología. El tratamiento de los granulomas consiste en prevenir su formación, retirando el tubo lo antes posible o administrando ciclos de corticoides previamente a la extubación. Los quistes subglóticos deben ser escindidos mediante láser CO2 o disección; mientras que, el tratamiento quirúrgico de la estenosis será siempre individualizado y la elección de la técnica quirúrgica dependerá de la localización, grado de obstrucción, extensión y estado de la función laríngea.
Obstrucción laríngea por reflujo gastroesofágico
El reflujo gastro-esofágico-laríngeo (RGEL) se define como la presencia de ácido procedente del estómago en la porción más proximal del complejo laringofaríngeo. Se le considera como uno de los agentes etiológicos más destacado en casos de disfonía crónica, estenosis laringotraqueal, granulomas y laringitis posterior. Aunque la laringoscopia indirecta y la endoscopia son indicativas, la pH-metría de 24 horas, con doble o triple canal, se considera la prueba de elección para determinar el verdadero factor etiológico. En la práctica clínica, valorar la respuesta al tratamiento médico también constituye una prueba diagnóstica de gran utilidad. Debe tratarse el RGEL y valorar la evolución, antes de realizar cualquier procedimiento invasivo en estos niños(5,6).
Papilomatosis laríngea infantil
La papilomatosis laríngea es una enfermedad de origen viral, causada por el virus del papiloma humano y, principalmente, por los serotipos 6 y 11. Constituye la tumoración laríngea benigna más frecuente en los niños y es la segunda causa de estridor en la infancia.
Su incidencia varía en función de los autores, oscilando entre 3,6 y 4,3 casos por cada 100.000 niños. Puede presentarse a cualquier edad, aunque la incidencia es mayor entre los 2 y los 4 años.
La disfonía progresiva o la presencia de un lloro anómalo suelen ser signos tempranos de esta enfermedad. En las formas muy proliferativas, el paciente puede presentar disnea, observándose un estridor de inicio gradual y progresivo durante semanas o meses. La disfunción respiratoria aguda es rara.
El curso de la enfermedad es imprevisible; de manera que, puede responder al tratamiento, recurrir de forma crónica o retroceder espontáneamente al cabo de algunos años. A pesar de ello, la recidiva es la norma general. El riesgo de malignización en niños es extremadamente bajo.
La papilomatosis laríngea constituye una entidad clínica de difícil tratamiento, debido a las recurrencias y complicaciones asociadas. Actualmente, el tratamiento de elección es la vaporización con láser CO2. La media de intervenciones en la papilomatosis infantil es de 4 intervenciones por año, con una eliminación total de las lesiones superior al 50%. También pueden utilizarse terapias coadyuvantes, como el interferón alfa, enzimas proteolíticas o inhibidores del crecimiento tumoral (cidofovir), que se han demostrado útiles para reducir el tamaño tumoral y alargar el tiempo entre las recidivas(8).
Tuberculosis laríngea
Suele cursar con una clínica de faringitis asociada a lesiones membranosas y ulceraciones mucosas. No suele asociarse con afectación pulmonar. El diagnóstico se realiza mediante endoscopia y es necesario realizar biopsias para identificar el Mycobacterium tuberculosis mediante cultivo o identificación genética. Se utilizan las pautas de tratamiento clásico(2).
Laringomalacia
La laringomalacia se define como el colapso intraluminal de las estructuras supraglóticas durante la inspiración. Constituye la causa más frecuente de estridor en la infancia (65-75%), siendo, además, la anomalía congénita más habitual de la laringe. El mecanismo etiopatogénico es desconocido, pero se han descrito diferentes factores anatómicos, histológicos y neurológicos, que pueden influir en el desarrollo de esta patología.
El estridor, inspiratorio e intermitente, es la manifestación clínica típica de la laringomalacia. El pronóstico es favorable y, en la mayoría de casos (70%), se produce una mejoría espontánea y gradual antes de los 12-24 meses de vida. Sin embargo, en el 10% de los casos, la obstrucción es severa y puede producir: cianosis, apnea, insuficiencia cardiaca e hipertensión pulmonar, siendo necesaria la cirugía para su tratamiento. La laringomalacia puede presentarse de forma aislada o asociada a otras anomalías de la vía respiratoria (19%) u otros órganos sistémicos.
Para el diagnóstico, es muy importante la historia clínica, valorar la existencia de estridor, sus características y la presencia o no de síntomas asociados. El diagnóstico definitivo se establece mediante endoscopia. La laringe debe visualizarse con respiración espontánea para valorar de forma dinámica su función y morfología.
El tratamiento inicial es expectante; ya que, está demostrado que, en la mayoría de casos, los síntomas desaparecen espontáneamente antes de los dos años de vida. El tratamiento quirúrgico (epiglotoplastia/traqueotomía) se reserva para aquellos casos con síntomas severos y repercusión orgánica grave(9).
Parálisis congénita de cuerdas vocales
Constituye el 10% de las anomalías congénitas. La parálisis unilateral se asocia a causas periféricas, siendo la más frecuente el estiramiento del cuello durante el parto. El pronóstico es bueno y suele curar de forma espontánea.
Las parálisis bilaterales casi siempre se relacionan con problemas del sistema nervioso central. Tienen peor pronóstico; se recomienda traqueotomía y esperar evolución.
Estenosis subglótica congénita
Es la tercera anomalía congénita por orden de frecuencia. Se admiten como estenosis todas aquellas en las que se objetiva, mediante endoscopia, un diámetro subglótico inferior al correspondiente para la edad y el peso. Este tipo de estenosis suele mejorar espontáneamente con el crecimiento. La traqueotomía y/o laringoplastia sólo se precisan en casos muy graves(2).
Hemangioma subglótico
Coincide con hemangiomas cutáneos en el 50% de los casos. Predominancia femenina. Generalmente, el tratamiento es conservador, ya que suelen regresar de forma espontánea antes de los dos años. El tratamiento quirúrgico es excepcional.
Rinofaringe y orofaringe
El segundo tramo de las vías aéreas superiores lo comprenden la rinofaringe y la orofaringe.
Fundamentalmente, la obstrucción de estas dos áreas tiene dos causas fundamentales: infamatorias y tumorales.
Inflamatorias
La obstrucción más frecuente de esta zona la provoca la hipertrofia del tejido linfoide situado a este nivel, las vegetaciones adenoideas en la rinofaringe y las amígdalas palatinas en la orofaringe (ambas estructuras forman parte del anillo de Waldeyer, que se complementa con los rodetes tubáricos y la amígdala lingual).
Las vegetaciones adenoideas son pliegues de mucosa respiratoria ubicados en la rinofaringe. Las amígdalas palatinas son auténticos ganglios linfáticos con un importante centro germinal. A diferencia de las primeras, en vez de pliegues, en su superficie tienen criptas.
Estas estructuras se ven con frecuencia afectadas de forma recurrente por cuadros virales o bacterianos, ambientes polucionados e irritativos e, incluso, por alergias alimentarias. La patología adenoamigdalar representa uno de los motivos más frecuentes de consulta médica (más de 4 millones de consultas al año en España). Las infecciones adenoamigdalares no sólo son importantes por sí solas, sino por la repercusión que tienen en las estructuras vecinas: nariz, senos paranasales, oído medio y vía aérea superior, cuya obstrucción nos ocupa y puede tener consecuencias sistémicas. La infección y la obstrucción coexisten con frecuencia.
Cierto grado de hipertrofia adenoidea se considera fisiológico hasta los cinco años. El diagnóstico de la hipertrofia adenoidea es clínico; ya que la radiografía lateral de cavum para el diagnóstico está en desuso. En pacientes colaboradores, el diagnóstico es más preciso con visión endoscópica directa(10).
Clínicamente, la hipertrofia adenoidea se manifiesta por obstrucción nasal crónica y ronquido (raramente se asocia a apneas nocturnas en ausencia de hipertrofia amigdalar acompañante). La inflamación crónica o aguda recurrente determina la aparición de otitis media. En casos de larga evolución, se han descrito alteraciones del crecimiento facial. La hipertrofia amigdalar es la principal causa de obstrucción durante el sueño.
El fenómeno obstructivo más llamativo de este área se produce, pues, principalmente durante el sueño y su importancia es de tal magnitud que merece una atención especial:
Los Trastornos Respiratorios del Sueño (TRS) en la infancia deben considerarse como una entidad diferenciada del cuadro clínico del adulto, al margen de que existen aspectos que comparten. Las diferencias se observan en todos los capítulos de la enfermedad. Las alteraciones anatómicas en los TRS infantil tienen mayor preponderancia y existen también diferencias en sus manifestaciones clínicas. No existen muchos datos epidemiológicos sobre la enfermedad en niños. Los estudios bien conformados en adultos ya son escasos y complicados de realizar y en niños estas dificultades se incrementan al intentar aplicar los criterios diagnósticos del adulto. Aproximadamente, el 10-12% de los niños roncan y muchos de ellos tienen el mal llamado ronquido simple, que es el ronquido que ocurre sin asociarse a apnea o a alteraciones del intercambio gaseoso(11). Su frecuencia declina a partir de los 9 años. La prevalencia de apnea del sueño en niños se estima entre el 0,7% y el 3%, con un pico entre los 2 y 5 años, y la del ronquido se sitúa entre un 7 y un 9%. En los TRS tienen influencia factores ambientales, factores del desarrollo del niño, así como múltiples genes. Los genes específicos que influyen en el desarrollo de esta patología no han sido determinados, aunque se tiene cada vez más evidencia de la existencia de una base genética en su expresión. Muchos estudios han demostrado ya la presencia de una agregación familiar. Las características fenotípicas del individuo, así como determinados genes relacionados con los principales factores de riesgo y mediadores biológicos relacionados con la expresión clínica influyen en los mecanismos celulares y moleculares implicados en su etiopatogenia(12).
Fisiopatología
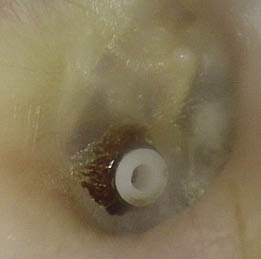
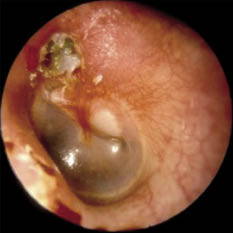
Los estudios actuales sobre la fisiopatología van más dirigidos a alteraciones de función (alteración muscular o de control neural) que no a alteraciones anatómicas que comporten obstrucción. Sin embargo, en la población infantil, el factor anatómico es muy evidente. Además, existen factores inherentes a su anatomía que facilitan el colapso: vías aéreas altas más estrechas, posición más alta de la laringe, mayor laxitud de los tejidos y mayor flexibilidad cartilaginosa de las estructuras de soporte. Los casos más frecuentes de TRS infantil son los derivados de las hipertrofias adenoamigdalares. Suponen tres cuartas partes de casos en la mayoría de series (Fig. 4). Además, los procesos infecciosos de vías altas repetidos pueden agravar TRS leves o desencadenar su desarrollo en niños. Además de esta causa, se puede observar TRS en alteraciones craneofaciales, como las de los síndromes de Apert, Crouzon o Treacher-Collins (hipoplasia maxilar y descenso del diámetro anteroposterior de faringe), en el síndrome de Pierre-Robin (micrognatia, glosoptosis, hendidura palatina e hipotonía muscular), en el síndrome de Down o acondroplasia (alteraciones faciales y macroglosia) o en las mucopolisacaridosis (hipertrofia del tejido linfoide faríngeo). Se ha sugerido una base familiar del TRS, probablemente por disfunciones del control ventilatorio o de la morfología cráneo-facial, que pueden heredarse(12). El modo respiratorio y la morfología craneofacial están estrechamente conectados. La hipertrofia adenoidea o amigdalar pueden influir en el desarrollo de la hemicara inferior y mandibular, al provocar respiración oral. La respiración oral en el niño es un hecho muy común en la infancia, afecta al 25% de los menores de 10 años. Todo ello conlleva una modificación en el patrón de crecimiento, con planos faciales divergentes y arcos dentales estrechos. La persistencia de este cuadro, junto con sus factores desencadenantes, perpetúa el proceso de respiración oral, generando un círculo vicioso que influye negativamente en el desarrollo esquelético facial y de estructuras internas, como la vía aérea superior.
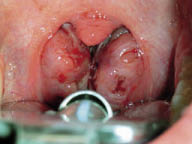
Figura 4. Hipertofia amigdalar obstructiva.
Clínica y consecuencias negativas
Durante el sueño, los síntomas más relevantes son el ronquido o la respiración ruidosa. También pueden constatarse: pausas respiratorias, sueño intranquilo con posturas anómalas, enuresis, sudoración abundante o la existencia de pesadillas o terrores nocturnos. También, es frecuente la presencia de bruxismo o, incluso, sonambulismo. En vigilia, al contrario que en el adulto, la somnolencia no es frecuente, pero sí los trastornos del comportamiento: hiperactividad, déficit de atención, escaso rendimiento escolar, inhibición social, mal carácter o agresividad. Puede existir cefalea matutina, sed excesiva al levantarse o síntomas derivados de la hipertrofia adenoamigdalar, como respiración oral o disfagia(13). Una consecuencia negativa importante de los TRS son las alteraciones del comportamiento y del desarrollo neurocognitivo. En estos niños se multiplica por tres la frecuencia de alteraciones de conducta y problemas de atención. Estos parámetros son importantes, ya que la capacidad de atención juega un papel importante en el aprendizaje y, por tanto, en desarrollo social y académico. Las diferentes alteraciones publicadas en niños con apnea del sueño en relación al campo del conocimiento van desde alteraciones en inteligencia general y memoria, hasta inteligencia verbal y alteraciones cognitivas ejecutivas. Es importante el rápido reconocimiento y tratamiento de las alteraciones del sueño, dado que suceden en un momento de la vida (primera década) de rápido desarrollo neurocongnitivo. Los TRS pueden asociarse con retraso en el desarrollo pondoestatural. Entre el 4 y el 13% de los niños con trastornos respiratorios del sueño tienen problemas de crecimiento. Este retraso en el desarrollo se ha atribuido, sobre todo, a un excesivo consumo energético nocturno por parte de la musculatura respiratoria y a que la alteración de la arquitectura del sueño interfiere en la liberación de la hormona del crecimiento que se produce, sobre todo, en la fase de ondas lentas. Otras posibles causas son la anorexia o disfagia debidas a la hipertrofia adenoamigdalar y a la hipoxemia y acidosis respiratoria, nocturnas. Los niveles de IGF-I e IGFBP-3, parámetros que dependen de la hormona de crecimiento, experimentan un incremento significativo tras el tratamiento de la enfermedad, con una normalización de la secreción de GH. Otra posible consecuencia negativa son las alteraciones cardiovasculares. Existe evidencia de hipertrofia ventricular derecha en un 55% de los niños con SAOS. La apnea del sueño expone a la población infantil a hipoxemia intermitente nocturna y estrés oxidativo. Estos factores amplifican el efecto de la adiposidad sobre el estado inflamatorio sistémico y favorecen el desarrollo de alteraciones metabólicas que derivan en diabetes y enfermedades vasculares. La obstrucción de la vía aérea superior comporta apnea e hipoxia durante el sueño y puede progresar hacia un incremento de la presión arterial pulmonar y, en algunos casos, a cor pulmonale. A este efecto de presión debe añadirse la posibilidad de que el incremento de resistencias esté favorecido por la hiperviscosidad. La presencia de HTA es muy frecuente en el caso del adulto, pero en el niño, aparentemente, no tanto y lo mismo sucede con las alteraciones del ritmo. Sin embargo, se ha demostrado que los niños con un IAH (índice de apnea-hipoapnea) mayor de 10 exhiben presiones diastólicas y sistólicas mayores que los casos con IAH inferiores a 10.
Diagnóstico
La historia clínica y la exploración física no son capaces, por sí solas, de diferenciar el roncador primario del niño con apneas nocturnas. La polisomnografía nocturna sigue siendo el mejor método, aunque los problemas derivados de su alto coste y de la necesidad, sobre todo en niños, de personal adiestrado, siguen provocando el intento de ofertar métodos alternativos. Los episodios de apnea obstructiva completa suelen faltar en los niños con TRS. Los criterios de adulto basados en el IAH fallan a la hora de identificar a niños con TRS y son más importantes los criterios gasométricos y los basados en los esfuerzos respiratorios paradójicos, para evaluar la gravedad del TRS en los niños. El diagnóstico anatómico o de localización de la causa obstructiva es más importante en la población infantil, dado que, en el niño, estos factores tienen más protagonismo. Se pueden apreciar con facilidad las alteraciones anatómicas que condicionan en tantos casos el TRS: hipertrofia adenoamigdalar, alteraciones craneofaciales y trastornos neuromusculares(12). En la exploración física, no es frecuente hallar la obesidad tan típica en los casos de adulto. La prevalencia de la obesidad infantil en España está en aumento. Aproximadamente, un tercio de los niños con sobrepeso tienen TRS. La obesidad en niños con apnea del sueño oscila entre el 13 y el 36%. Se ha demostrado que, en los obesos, predominan claramente, y de forma significativa, las alteraciones respiratorias nocturnas en frecuencia y severidad. Por otro lado, los principales problemas de la adenoamigdalectomía, la persistencia del cuadro y las complicaciones postoperatorias, son más prevalentes en obesos. Por ello, es importante conocerlo para reducir peso previamente o bien prever estas eventualidades post-quirúrgicas en este grupo de niños. Radiológicamente, la placa simple lateral de partes blandas de cuello puede mostrar, con relativa fiabilidad, el tamaño de las vegetaciones adenoideas, aunque difícilmente predice el grado de obstrucción real y menos durante el sueño. Más importante es el estudio cefalométrico en los niños con alteraciones craneomandibulares (Fig. 5). Otras técnicas de imagen, como: fluoroscopia, resonancia nuclear magnética, tomografía axial computarizada ultrarrápida o cine-TC pueden ser útiles para casos concretos e investigación, pero no suelen aplicarse en la práctica diaria. La fibroendoscopia de la vía aérea superior forma parte del diagnóstico de localización de la obstrucción, a pesar de que, como todas estas técnicas no reproduce, en realidad, lo que sucede durante el sueño. Con ella, pueden evaluarse con precisión los tres niveles de la exploración endoscópica de la vía aérea: rinofaringe, orofaringe e hipofaringe.

Figura 5. Micrognatia
Tratamiento
La adenoamigdalectomía es el tratamiento más extendido y eficaz en casi tres cuartas partes de los casos. Consigue la normalización del cuadro respiratorio nocturno, de la sintomatología diurna y la reversión, en muchos casos, de las complicaciones cardiovasculares y del retraso en el crecimiento. En el momento actual, a la mayoría de niños a los que se indica la amigdalectomía, es debido a la existencia de una obstrucción de la vía aérea. El síndrome de apneas obstructivas del sueño es, pues, la indicación más frecuente de amigdalectomía. Habitualmente, debe asociarse a adenoidectomía en el mismo acto quirúrgico(12,14).
Amigdalectomía. Técnicas quirúrgicas
Se han descrito multitud de técnicas de amigdalectomía. Los procedimientos con el paciente despierto (guillotina de Sluder) son inaceptables hoy día debido al alto riesgo de complicaciones. La disección bajo anestesia general e intubación orotraqueal es el procedimiento más habitual. Se han descrito múltiples modificaciones de la técnica de disección (disección fría, bipolar, láser, criocirugía, radiofrecuencia, etc.), que no han disminuido significativamente la morbilidad de la intervención, ni la tasa de complicaciones.
• Técnica de amigdalectomía por disección fría. El procedimiento universalmente admitido es la técnica de disección. Esta técnica se realiza en un quirófano correctamente equipado y bajo anestesia general con intubación oro-traqueal. Con el paciente en decúbito supino, el cirujano se coloca detrás de la cabeza y, con la ayuda de un abrebocas que deprime la lengua, se procede a la disección extracapsular de las amígdalas.
• Amigdalectomía por electrodisección. El electrobisturí y el electrocauterio se utilizan en cirugía para muchas intervenciones. Se consigue la hemostasia del campo quirúrgico más fácilmente que con otras técnicas y se acorta el tiempo operatorio. Se puede realizar con bisturí monopolar o bipolar ayudada o no con microscopio. Con bisturí monopolar el dolor es superior a la disección “fría”.
• Radiofrecuencia. Consiste en la generación de calor por medio de radiación electromagnética. La radiofrecuencia se puede utilizar como instrumento de disección para realizar la amigdalectomía total extracapsular (similar a la disección fría y a la electrodisección), como bisturí para realizar amigdalotomía (o amigdalectomía parcial) o como reductor del volumen amigdalar por medio de una sonda de radiofrecuencia en el interior de la amígdala. Con la radiofrecuencia, se consiguen temperaturas tisulares más bajas que con el láser o electrocauterio, con un menor daño térmico tisular, lo que ocasiona menos dolor postoperatorio que la electrodisección.
• Láser CO2. El láser CO2 puede utilizarse como instrumento para la disección extracapsular (amigdalectomía total) o como bisturí para amigdalotomía (amigdalectomía parcial):
– Amigdalectomía total (disección extracapsular): no posee ventajas comprobadas y sí un alto costo. Es una técnica lenta, por lo que está reservada sólo para aquellos pacientes con patologías tales como hemofilia u otros trastornos importantes de la coagulación(16-19).
Debido a las altas temperaturas tisulares, se produce más dolor que con técnicas tradicionales y no reduce la hemorragia postoperatoria comparada con otras técnicas.
– Amigdalectomía parcial (amigdalotomía): es una alternativa razonable para la hipertrofia amigdalar obstructiva. Junto a la amigdalotomía con radiofrecuencia, es la técnica con menor morbilidad (mínimo sangrado intra y postoperatorio y escaso dolor postoperatorio), debido a la ausencia de desgarro de la cápsula amigdalina(2,15).
Existen, sin embargo, otras propuestas terapéuticas. La postura y la dieta pueden ser elementos importantes en la estrategia terapéutica. La reducción ponderal en niños, por sí sola, no suele ser suficiente para solucionar el problema. Un metaanálisis reciente señala que la gravedad del TRS se relaciona con el grado de obesidad y que los niños con apnea del sueño y obesos tienen proporcionalmente más HTA. Sabemos que el tratamiento estrella de esta patología es la cirugía adenoamigadalar; se ha señalado que los de los principales problemas de esta cirugía, la persistencia del cuadro y las complicaciones postoperatorias son más prevalentes en obesos. La posición en decúbito supino es altamente conocido que tiene una influencia negativa en el caso del adulto. En niños no está tan claro, sin embargo y pensando en la relación con el síndrome de muerte súbita, parece ser aceptado, que la posición en decúbito prono es más peligrosa. Existen evidencias de que, en los niños, la colapsabilidad de la vía aérea superior aumenta en decúbito prono. El tratamiento médico de los TRS en los niños no es el tratamiento de elección. Existen tratamientos publicados, casi de forma anecdótica, como la insuflación faríngea con oxígeno durante el sueño o el tratamiento antiinflamatorio con antileucotrienos (Montelukast) que parecen reducir el volumen de las estructuras linfoides hipertrofiadas. Sí tienen mayor relevancia los tratamientos que mejoran la permeabilidad nasal. La obstrucción nasal o nasofaríngea colabora en el colapso faríngeo durante el sueño y su resolución, al igual que sucede en el adulto, podría ser un tratamiento, al menos, coadyuvante. Los tratamientos ortopédicos maxilares(2,14), además de conseguir una mejoría estética y oclusal, inducen variaciones esqueléticas que llevan a un cambio significativo en las estructuras internas de soporte de la vía aérea superior (VAS). La ortopedia del maxilar superior es capaz de producir una disminución de las resistencias nasales, mejorar la postura natural de la cabeza y aumentar la VAS. Además, induce un cambio en la dirección de crecimiento mandibular que conduce a una mejora en las proporciones esqueléticas faciales y un cambio en los lugares de inserción muscular encargados de garantizar la permeabilidad de la VAS. Debemos entender estas formas de corrección ortopédica como un mecanismo de profilaxis frente a una futura patología obstructiva de la VAS en el adulto. La cirugía sobre el macizo maxilar y las distracciones mandibulares pueden aumentar los espacios naso y orofaríngeos, mejorando las resistencias en el paso aéreo en los pacientes con TRS. Las estrategias terapéuticas, sobre todo en los casos de malformaciones, deben ser individualizadas a cada caso(14). En los casos de cirugía mayor maxilofacial para estas malformaciones, a pesar de corregirlas correctamente, no siempre consiguen solucionar el problema. La glosopexia, utilizada, sobre todo, en casos de Pierre-Robin, los avances maxilares y correcciones de anomalías congénitas en niños con síndrome de Crouzon o Apert, siempre con una correcta selección de los casos y con la experiencia necesaria pueden ser útiles. Las series publicadas no son amplias y muchas describen técnicas de expansión mandibular, ya sea mediante aparatos intraorales que la provocan de forma progresiva mediante un tornillo en el eje necesario (Rapid Maxillary Expansion) o bien mediante maniobras de distracción mandibular con osteogenésis secundaria. En los últimos años, el tratamiento con presión nasal positiva continua (CPAP) se ha convertido en la segunda línea de tratamiento cuando la apnea obstructiva persiste a pesar de la intervención quirúrgica(12). Otras indicaciones estarían destinadas al periodo prequirúrgico para estabilizar a niños con riesgo de compromiso posquirúrgico o en el tiempo necesario de crecimiento craneofacial hasta la cirugía definitiva. Existen diversos problemas al respecto que se revisarán en este capítulo. El cálculo del nivel de presión necesaria varía con el crecimiento y, por tanto, se hacen necesarias revisiones periódicas con polisomnografía. Existe una mayor tasa de intolerancia e incumplimiento a estas edades. Se requiere personal de soporte, como enfermeras entrenadas y la colaboración de los padres para conseguir la máxima aceptación por parte del niño. Finalmente, la estandarización de mascarillas especiales, sobre todo en niños con alteraciones craneofaciales que requieren diseños individuales, puede suponer otro problema. Además de la obstrucción de la vía respiratoria, la amigdalectomía tiene otras indicaciones:
• Amigdalitis agudas recurrentes.
• Disfagia mecánica, por hipertrofia amigdalar obstructiva de la orofaringe.
• Absceso periamigdalino. En su primer episodio o en el segundo, según los autores.
• Amigdalitis crónica resistente al tratamiento, en la que se producirían microabscesos en las criptas amigdalares que darían lugar a febrícula crónica y manifestaciones infecciosas generales(10).
Adenoidectomía
La adenoidectomía se debe realizar siempre en quirófano bajo anestesia general con intubación. Con el paciente en decúbito supino se coloca un abrebocas estático que sujeta la lengua. Con un instrumento de corte, diseñado para esta finalidad, se practica un legrado del cavum rinofaríngeo.
Las indicaciones de adenoidectomía son:
• Obstrucción nasal crónica diurna de más de 6 meses de evolución.
• Síndrome de apnea del sueño (asociada a amigdalectomía).
• Otitis media recurrente.
• Rinosinusitis aguda recurrente o crónica.
• Otitis serosa crónica.
Tumorales
Tumores benignos(2)
Angiofibroma de cavum. Es una tumoración que predomina en adolescentes varones. Su diagnóstico precoz es muy importante, pues de su extensión dependerá la agresividad y complejidad del tratamiento. No es lo mismo la extirpación de un tumor que afecte solamente a nasofaringe y senos, a que se extienda hacia la fosa ptérigo-maxilar, al suelo de la órbita o a la fosa infratemporal y que termine con una extensión intracraneal. El diagnóstico por excelencia se realiza mediante la RM y es imperativo no practicar una biopsia de una tumoración de cavum, sin haberla hecho antes, pues corremos el riesgo de provocar una hemorragia cataclísmica.
Su tratamiento es siempre quirúrgico. Es recomendable embolizarlo previamente y preparar sangre para una autotransfusión.
Tumores malignos(2,7)
• El carcinoma de nasofaringe es el más frecuente en la adolescencia. En el 60% de los casos, su primera manifestación es una adenopatía cervical y es la endoscopia nasal la que nos muestra una masa localizada en la pared posterior o externa de la rinofaringe.
Después del consecuente estudio por imagen, se diagnostica mediante biopsia. Ésta se debe realizar en quirófano, ya que muchas veces existe en la superficie tejido inflamatorio de aspecto polipoide o tejido necrótico que enmascaran el tumor. En los niños, predomina el carcinoma no queratinizante indiferenciado (linfoepitelioma), que tiene tendencia a metastatizar. Es un tumor en el que existe la participación del virus de Epstein-Barr.
Se trata con radioterapia, pudiéndose complementar con quimioterapia en caso de que haya metástasis. Responde bien a la radioterapia con una supervivencia del 60% a los 5 años. Sin embargo, entre el 20 y el 60%, pueden metastatizar (principalmente en pulmón, hígado y huesos).
• En nasofaringe y amígdalas podemos también encontrar linfomas MALT (tejido linfoide asociado a mucosas). Están compuestos por células linfocitarias B, de la zona marginal del folículo linfoide. También podemos encontrar localizado en la rinofaringe el linfoma no Burkitt.
Bibliografía
Los asteriscos reflejan el interés del artículo a juicio del autor.
1.** González A. Malformaciones congénitas y deformaciones de la nariz, fosa y senos paranasales. En: Tratado de Otorrinolaringología y Cirugía de Cabeza y Cuello. Tomo I. Ciencias básicas y materias afines. Rinología. Madrid: Editorial Proyectos Médicos; 1999. p. 457-74.
2.*** Tomás M, Bernal M. Tratado de Otorrinolaringología Pediátrica. Girona: Gráficas Alzamora; 2000 (Ponencia oficial de la SEORL 2000).
3.* Fernández Pérez AJ, Burgos Sánchez AJ, Gras Albert JR. Lesiones congénitas de la línea media nasofrontal. Acta Otorrinolarig Esp. 2001; 52: 404-8.
4.** Massegur Solench H, Ademà Alcover JM. Rinitis y sinusitis en el niño. En: Formación continuada de la Sociedad Española de Otorrinolaringología y Patología Cérvico-Facial. Vol II. Infección en ORL: Barcelona: Editorial Masson; 2005. p. 1-10.
5.** Rosbe KW, Kenna MA, Auerbach AD, Extraesophageal reflux in Pediatric Patients With Upper Respiratory Symptoms. Arch Otol Head Neck Surg. 2003; 129: 1213-20.
6.*** Yellon RF, Goldber H. Update on Gastroesophageal Reflux Disease in Pediatric Airway disorders. Am J Med. 2001; 111(suppl 8A): 78S-84S.
7.*** Muñoz Borge F, González Alonso J, Galera Ruiz H, Delgado Moreno F, Galera Davidson H. Avances en los tumores Otorrinoloringológicos. An Pediatr. 2003; 58(5) 456-63.
8.** Derkay Craig S. Recurrent Respiratory Papillomatosis. Laryngoscope. 2001; 111; 57-69.
9.** Olney DR, Greinwald JH Jr, Smith RJ, Bauman NM. Laryngomalacia and its Treatment. Laryngoscope. 2001; 109: 1770-5.
10.* Benito Jiménez J, Villafruela Sanz MA. Vegetaciones adenoideas. Fisiopatología. Indicaciones de tratamiento. Otitis media con efusión. An Pediatr Monogr. 2003; 1(1): 72-80.
11.** Owen G, Canter R, Maw R. Screening for obstructive sleep apnoea in children Int J Pediatr Otorhinolaryngol. 1995; 32(Suppl): s67-9.
12.*** Guilleminault C, Pelayo R. Sleep-disordered breathing in children. Ann Med. 1998; 4: 350-6.
13.** Gaultier C. Obstructive sleep apnea syndrome in infants and children: established facts and unsettled issues. Thorax. 1995; 50: 1204-10.
14.** Esteller E, Estivill E. El Ronquido y el Síndrome de la apnea obstructiva en los niños. Vigilia-Sueño. 2000; 12(1) (Supl): s29-s35.
15.*** Pinder D, Hilton M. Disección versus diatermia para la amigdalectomía (Revisión Cochrane traducida). En: La Biblioteca Cochrane Plus. 2005; Número 1.
16.* Llorente Pendas JL, Suárez C. Indicaciones de la adenoamigdalectomía. Boletín de la Sociedad de Pediatría de Asturias, Cantabria, Castilla y León. 1999; 39: 72-5.
17.** Melchor Díaz MA. Amigdalitis aguda. Criterios de amigdalectomía. An Pediatr Monogr. 2003; 1(1): 62-8.
18.** Nazar J. Amigdalectomía: nueva tecnología confrontada con la cirugía tradicional. Rev Otorrinolaringol Cir Cabeza Cuello. 2004; 64: 252-61.
19.*** Friedman M, Wilson M, Lin HC, Chang HW Updated systematic review of tonsillectomy and adenoidectomy for treatment of pediatric obstructive sleep apnea/hypopnea syndrome. Otolaryngol Head Neck Surg. 2009; 140(6): 800-8.
Bibliografía recomendada
– Guilleminault C, Pelayo R. Sleep-disordered breathing in children. Ann Med. 1998; 4: 350-6.
Importante revisión del SAOS infantil de un grupo de reconocido prestigio en el tema. Destacan los aspectos de predisposición familiar y los signos clínicos de la enfermedad en esta población. También, hacen referencia a los aspectos diferenciales con el cuadro clínico en adultos. Es importante que estos niños sean tratados mediante CPAP o cirugía para evitar complicaciones serias a largo plazo.
– Pinder D, Hilton M. Disección versus diatermia para la amigdalectomía (Revisión Cochrane traducida). En: La Biblioteca Cochrane Plus. 2005; Número 1.
Interesante artículo de revisión de ensayos aleatorios en niños y adultos sometidos a cirugía amigdalar en el que compara la morbilidad de la amigdalectomía realizada con dos técnicas diferentes, la disección fría y la electrodisección. Concluye que todavía no se ha demostrado claramente la superioridad de una técnica sobre otra.
– Yellon RF, Goldber H. Update on Gastroesophageal Reflux Disease in Pediatric Airway disorders. Am J Med. 2001; 111(suppl 8A): 78S-84S.
El reflujo gastroesofágico (RGE) se ha relacionado con la aparición de tos, apnea, estenosis laringotraqueal, laringitis posterior y laringomalacia. Muchos de estos síntomas mejoran con tratamiento para el RGE. El diagnóstico definitivo se establece mediante La pH-metría de 24 horas.
– Schechter MS; Section on Pediatric Pulmonology, Subcommittee on Obstructive Sleep Apnea Syndrome. Technical report: diagnosis and management of childhood obstructive sleep apnea syndrome. Pediatrics. 2002; 109(4): e69.
Documento de la Academia Americana de Neumología Pediátrica donde se revisan todos los aspectos actuales sobre el abordaje terapéutico y diagnóstico del síndrome de la apnea del dueño en los niños. Se trata de un documento de referencia mundial sobre las recomendaciones a seguir en diferentes aspectos de tratamiento y diagnóstico.
– Friedman M, Wilson M, Lin HC, Chang HW. Updated systematic review of tonsillectomy and adenoidectomy for treatment of pediatric obstructive sleep apnea/hypopnea syndrome. Otolaryngol Head Neck Surg. 2009; 140(6): 800-8.
Una revisión exhaustiva y sistemática sobre el tratamiento más extendido y eficaz de la apnea obstructiva en la infancia: la adenoamigdalectomía. Se revisan las técnicas, indicaciones y resultados de dicho tratamiento.
| Caso clínico |
|
Paciente de 3 años de edad que acude a la consulta del ORL por que los padres observan dificultad respiratoria nasal e intensos ronquidos nocturnos. El interrogatorio del ORL pone de manifiesto la presencia de apneas nocturnas prolongadas que asustan a la familia, enuresis nocturna y un sueño muy intranquilo. Profundizando más en la anamnesis, los padres refieren que el niño durante el día está muy irritable pero no excesivamente somnoliento.
En la exploración, se aprecia una hipertrofia amigdalar significativa y una cara larga con respiración oral continuada. En la exploración dental, se evidencia un paladar estrecho y una mordida abierta anterior. El registro polisomnográfico del niño muestra un índice de apnea-hipoapnea de 10,7 sin claras desaturaciones de O2. Se aconseja a los padres la realización de una reducción amigdalar con radiofrecuencia y adenoidectomía asociada bajo anestesia general y se realiza un estudio previo del riesgo quirúrgico con la colaboración de los Servicios de Anestesia y Pediatría. Se practica dicha intervención sin ningún tipo de incidencia peroperatoria ni postoperatoria y, a los diez días, el cuadro clínico del niño se resuelve por completo. El paciente es citado al cabo de un año para nuevo control clínico y polisomnográfico.
|